DADA2 Pipeline exemple du marqueur 16S
Louis Astorg
Juin 2023
- Mise en place de l’environnement R
- Suppression des amorces de séquences
- Inspection de la qualité
- Filtrer et rogner
- Génération d’un modèle d’erreur de nos données
- Déplication, inférences d’échantillons et fusion des séquences
- Construire un tableau de séquence
- Suppression des séquences chimères
- Suivi des séquences au travers du pipeline
- Attribuer une taxonomie
- Cleaning and saving files
- Éliminer les contaminants probables
- Clustering MOTUs
Mise en place de l’environnement R
Chargement des librairies
library(DECIPHER)
library(decontam)
library(dada2)
library(tidyverse)
Mise en place des fichiers
path_data <- "../data/"
path_raw_seq <- "../data/raw_sequences/"
path_cut <- "../data/cutadapt/"
path_doc <- "../docs/"
path_fig <- "../figs/"
path_out <- "../out/"
path_R <- "../R/"
path_scripts <- "../scripts/"
paths <- c(
path_data,
path_raw_seq,
path_cut,
path_doc,
path_fig,
path_out,
path_R,
path_scripts
)
for (i in 1:length(paths)) {
if (file.exists(paths[i])) {
cat(paste0("The folder ", i, " already exists\n"))
} else {
dir.create(paths[i])
}
}
Fonctions locales
source(paste0(path_R, "prep_cdm.R"))
Suppression des amorces de séquences
Cette partie est facultative. Si les amorces ont déjà été retirées, sautez-la.
Définition des patrons de fichiers
Ici, nous établissons le patron de fichier pour discriminer vos fichiers de séquences “forward” et “reverse”.
file_pattern <- c("F" = "_R1_001.fastq.gz",
"R" = "_R2_001.fastq.gz")
Définition des chemins d’accès aux fichiers
Cette étape définit les chemins d’accès à tous les fichiers de séquences brutes “forward” et “reverse”.
fas_Fs_raw <- paste0(path_raw_seq,
list.files(path_raw_seq,
pattern = file_pattern["F"]))
fas_Rs_raw <- paste0(path_raw_seq,
list.files(path_raw_seq,
pattern = file_pattern["R"]))
Définition des amorces de séquençage
Nous devons fournir les séquences d’amorces à partir desquelles nous pouvons également calculer le complément inverse des amorces.
FWD <- c("CCTACGGGAGGCAGCAG")
REV <- c("CTACCAGGGTATCTAATCC")
FWD_RC <- dada2:::rc(FWD)
REV_RC <- dada2:::rc(REV)
Définition des fichiers de sorties
Après l’opération de suppression de l’amorce, les fichiers traités doivent être envoyés dans un répertoire de notre choix.
fas_Fs_cut <- paste0(path_cut, basename(fas_Fs_raw))
fas_Rs_cut <- paste0(path_cut, basename(fas_Rs_raw))
Définition des arguments de base de cutadapt
Ici, nous précisons les arguments de la fonction cutadapt.
R1_flags <- paste(paste("-g", FWD, collapse = " "))
R2_flags <- paste(paste("-G", REV, collapse = " "))
Coupe des amorces avec cutadapt
Nous utilisons une boucle pour traiter chaque échantillon de manière itérative. L’argument discard-untrimed précise que nous voulons que les séquences non rognées soient rejetées car elles ne contiennent pas la séquence d’amorce. L’argument max-n 0 précise que l’on autorise 0 N paires de bases dans les séquences (si trop de séquences contiennent cette base, l’argument peut être modifié). N bases signifie que le logiciel Illumina n’a pas pu effectuer d’appel de base pour cette base.
# fichier contenant l'installation cutadapt à modifier !
cutadapt <- "/home/louis/anaconda3/envs/cutadapt/bin/cutadapt"
for(i in seq_along(fas_Fs_raw)) {
cat("Processing", "-----------", i, "/", length(fas_Fs_raw), "-----------\n")
system2(cutadapt,
args = c(R1_flags,
R2_flags,
"--discard-untrimmed",
"--max-n 0",
# Optional strong constraint on expected length
#paste0("-m ", 250-nchar(FWD)[1], ":", 250-nchar(REV)[1]),
#paste0("-M ", 250-nchar(FWD)[1], ":", 250-nchar(REV)[1]),
"-o", fas_Fs_cut[i], "-p", fas_Rs_cut[i],
fas_Fs_raw[i], fas_Rs_raw[i]))
}
Compiler le nombre de base
Nous créons un objet qui a le nombre de séquences de chaque échantillon avant et après avoir coupé les amorces. Si nous voyons une perte importante de séquences, quelque chose s’est probablement mal passé à cette étape et nous pourrons ajuster les contraintes de cutadapt.
out_1 <- cbind(ShortRead::qa(fas_Fs_raw)[["readCounts"]][,"read", drop = FALSE],
ShortRead::qa(fas_Fs_cut)[["readCounts"]][,"read", drop = FALSE])
out_1
read read
BLANK_S233_L001_R1_001.fastq.gz 27758 26837
sample-1_S213_L001_R1_001.fastq.gz 26844 26040
sample-2_S191_L001_R1_001.fastq.gz 34088 32788
sample-3_S223_L001_R1_001.fastq.gz 36464 35228
Inspection de la qualité
Définir des chemins des fichiers
Si vous avez ignoré la suppression des amorces, indiquez ici le chemin d’accès à vos séquences.
path_process <- path_cut # since we didn't skip primer removal
fas_Fs_process <- paste0(path_process,
list.files(path_raw_seq,
pattern = file_pattern["F"]))
fas_Rs_process <- paste0(path_process,
list.files(path_raw_seq,
pattern = file_pattern["R"]))
Noms des échantillons
Ici, nous créons un vecteur de noms d’échantillons propres que nous utiliserons dans les tables de sortie. Cette étape est très individuelle à vos données. Assurez-vous d’utiliser des noms faciles à transformer pour vos échantillons lorsque vous les envoyez au séquençage.
sample_names <- sapply(strsplit(basename(fas_Fs_process), "_"),
function(x) {paste0(x[c(1)],collapse="_")} )
Inspecter les profils de qualité des séquences
Nous créons un fichier pdf avec les profils de qualité des séquences moyennes des échantillons ou des 100 premiers échantillons s’il y en a plus de 100.
pdf(file.path(path_fig, "Read_quality_profile_aggregated.pdf"))
p1 <- plotQualityProfile(sample(fas_Fs_process,
replace = FALSE,
size = ifelse(length(fas_Fs_process) < 100,
length(fas_Fs_process), 100)),
aggregate = TRUE)
p1 + ggplot2::labs(title = "Forward")
p2 <- plotQualityProfile(sample(fas_Rs_process,
replace = FALSE,
size = ifelse(length(fas_Rs_process) < 100,
length(fas_Rs_process), 100)),
aggregate = TRUE)
p2+ ggplot2::labs(title = "Reverse")
dev.off()
quartz_off_screen
2
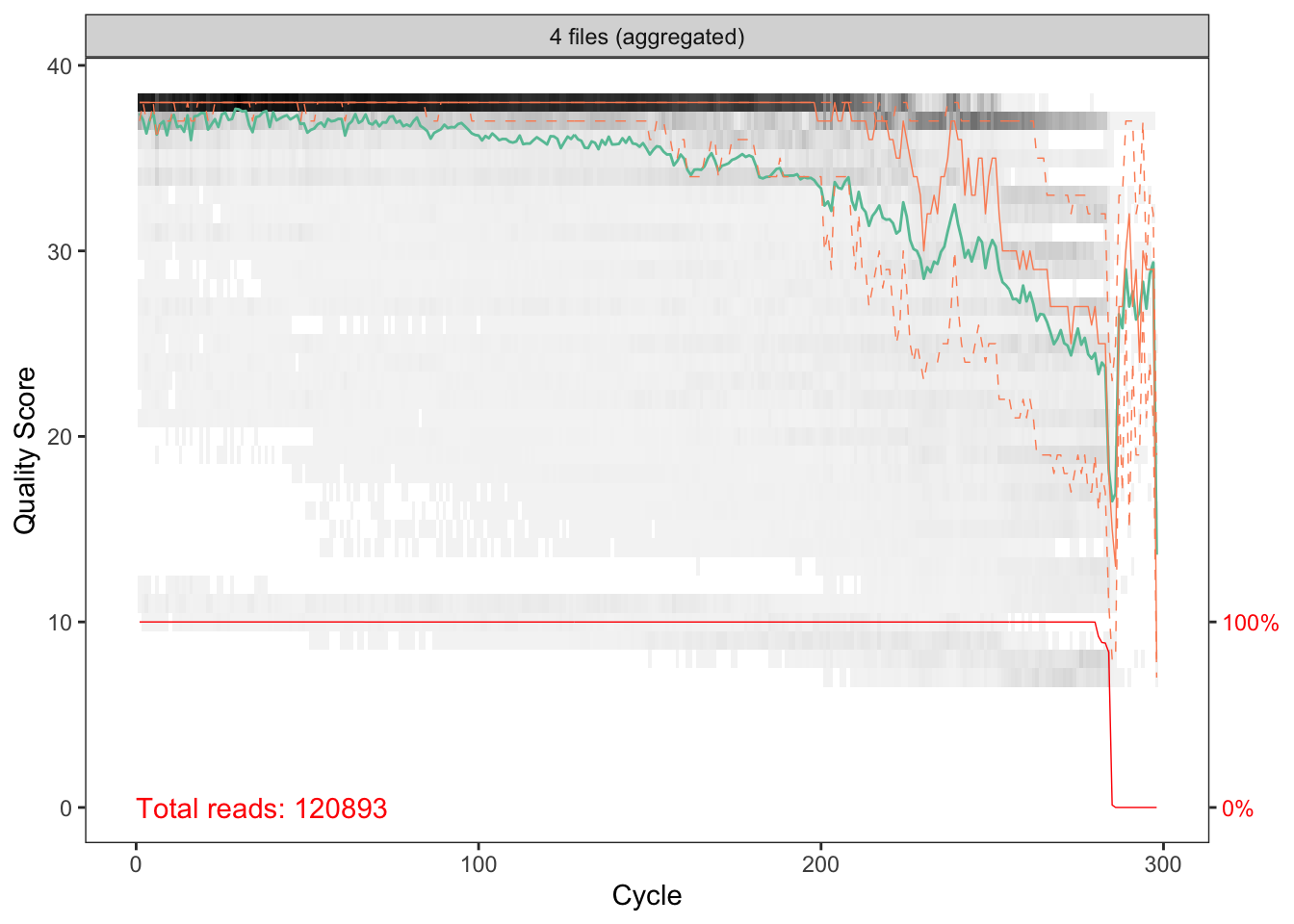
p1
p2
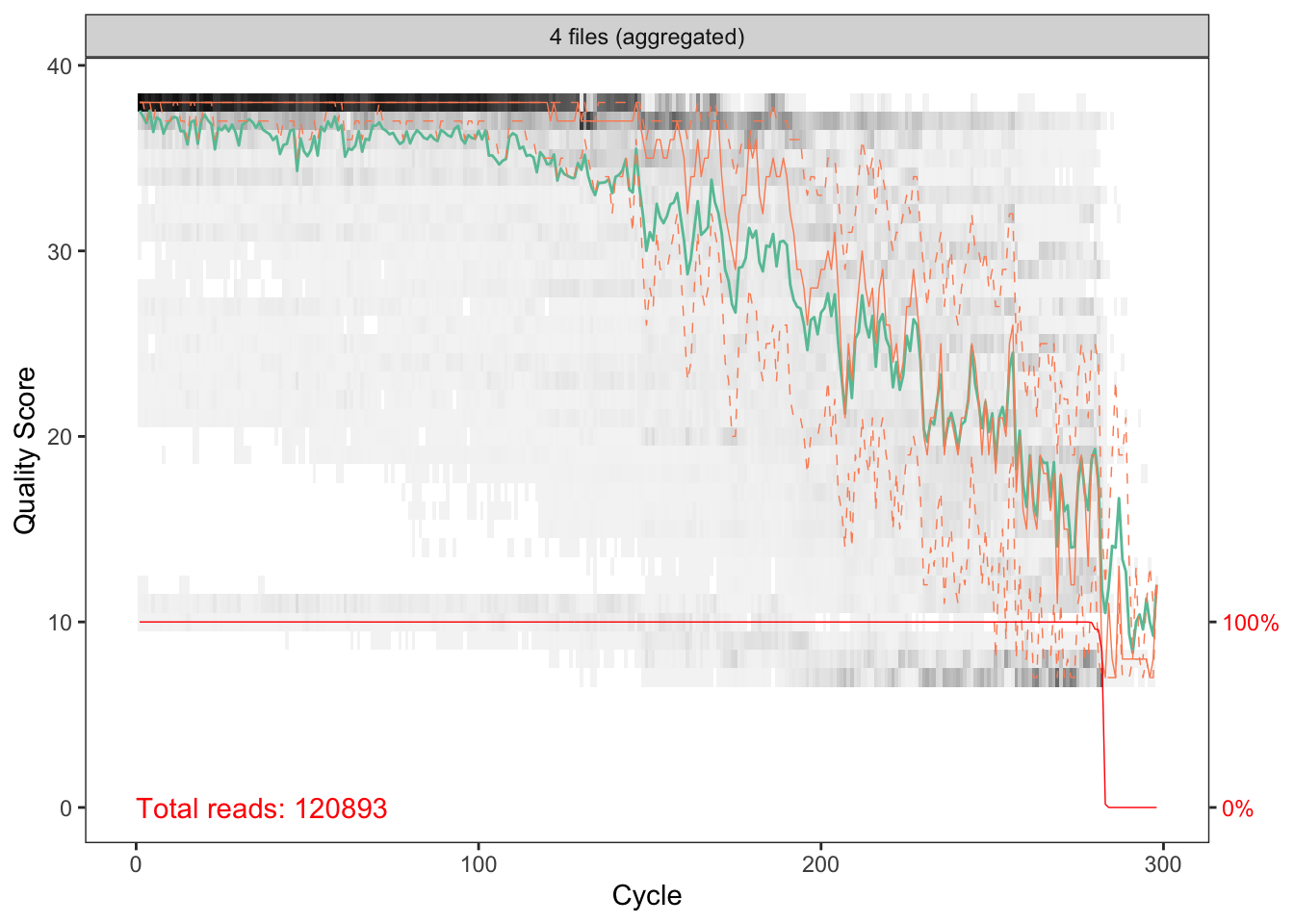
Sur ces graphiques, les bases sont le long de l’axe des x et le score de qualité sur l’axe des y. La heatmap sous-jacente noire montre la fréquence de chaque score à chaque position de base, la ligne verte est le score de qualité moyen à cette position de base, l’orange est la médiane et les lignes orange en pointillés montrent les quartiles. La ligne rouge en bas indique le pourcentage de lectures de cette longueur.
Filtrer et rogner
Définir des chemins des fichiers
Nous définissons un nouveau chemin pour les séquences filtrées. Nous vérifions également que les noms de fichiers sont toujours identiques aux séquences brutes.
fas_Fs_filtered <- paste0(path_data,
"filtered/",
basename(fas_Fs_process))
fas_Rs_filtered <- paste0(path_data,
"filtered/",
basename(fas_Rs_process))
all.equal(basename(fas_Fs_raw),
basename(fas_Fs_filtered))
Renomme les séquences avec des noms d’échantillons
Cette étape renomme les fichiers des séquences avec les noms d’échantillons en vue de la fusion des séquences “forward” et “reverse”.
names(fas_Fs_filtered) <- sample_names
names(fas_Rs_filtered) <- sample_names
Filtrage et découpage
Réfléchissez bien avant de copier la commande suivante! Idéalement, nous voulons couper les séquences pour garder une qualité supérieure à 30, mais il est également important de penser à vos amorces et au chevauchement que vous allez avoir. Si nous réduisont trop la taille des séqunces, il se pourrait que le recouvrement entre les séquences “forward” et “reverse” ne soit pas suffisant ou tout simplement absent. Cela causerait des problèmes plus tard dans le pipeline car nous ne pourrons pas fusionner ces séquences. Assurez-vous donc de bien prendre en compte la longueur des vos séquences et des vos amorces avant de couper. Ici, je vais couper les lectures avant à 220 et les lectures inverses à 200 – à peu près là où les deux ensembles maintiennent une qualité médiane de 30 ou plus – puis voir à quoi ressemblent les choses. Il existe également un paramètre de filtrage par défaut supplémentaire qui supprime toutes les séquences contenant des Ns, maxN, définis sur 0 par défaut. maxEE est le seuil de filtrage de qualité appliqué en fonction des erreurs attendues et dans ce cas, nous disons que nous voulons jeter la séauence si elle est susceptible d’avoir plus de 2 appels de base erronés. rm.phix supprime toutes les lectures qui correspondent au génome du bactériophage PhiX, qui est généralement ajouté aux séquences de séquençage Illumina pour le contrôle de la qualité.
out_2 <- filterAndTrim(fas_Fs_process,
fas_Fs_filtered,
fas_Rs_process,
fas_Rs_filtered,
truncLen = c(220,200),
maxN = 0,
maxEE = c(2, 2),
rm.phix = TRUE,
compress = TRUE,
multithread = TRUE)
head(out_2)
reads.in reads.out
BLANK_S233_L001_R1_001.fastq.gz 26837 20821
sample-1_S213_L001_R1_001.fastq.gz 26040 20685
sample-2_S191_L001_R1_001.fastq.gz 32788 26097
sample-3_S223_L001_R1_001.fastq.gz 35228 29093
Génération d’un modèle d’erreur de nos données
La prochaine étape consiste à générer notre modèle d’erreur avec la signature d’erreur spécifique de notre ensemble de données. Chaque cycle de séquençage, même lorsque tout se passe bien, aura ses propres variations dans son profil d’erreur. Cette étape tente d’évaluer ces profils d’erreur pour les séquences “forward” et “reverse”. Il s’agit de l’une des étapes les plus gourmandes en calcul du pipeline.
Calcul des modèles d’erreur
# error_F <- learnErrors(fas_Fs_filtered,
# multithread = TRUE,
# randomize = TRUE)
# error_R <- learnErrors(fas_Rs_filtered,
# multithread = TRUE,
# randomize = TRUE)
Si vous relancez l’analyse et que le modèle d’erreur est déjà enregistré, chargez-le ici.
load(paste0(path_R, "error_F.RData"))
load(paste0(path_R, "error_R.RData"))
Graphiques des modèles d’erreur
pdf(file.path(path_fig, "Error_rates_learning.pdf"))
pF <- plotErrors(error_F, nominalQ = TRUE)
pF + ggplot2::labs(title = "Error Forward")
pR <- plotErrors(error_R, nominalQ = TRUE)
pR + ggplot2::labs(title = "Error Reverse")
dev.off()
quartz_off_screen
2
pF + ggplot2::labs(title = "Error Forward")

pR + ggplot2::labs(title = "Error Reverse")
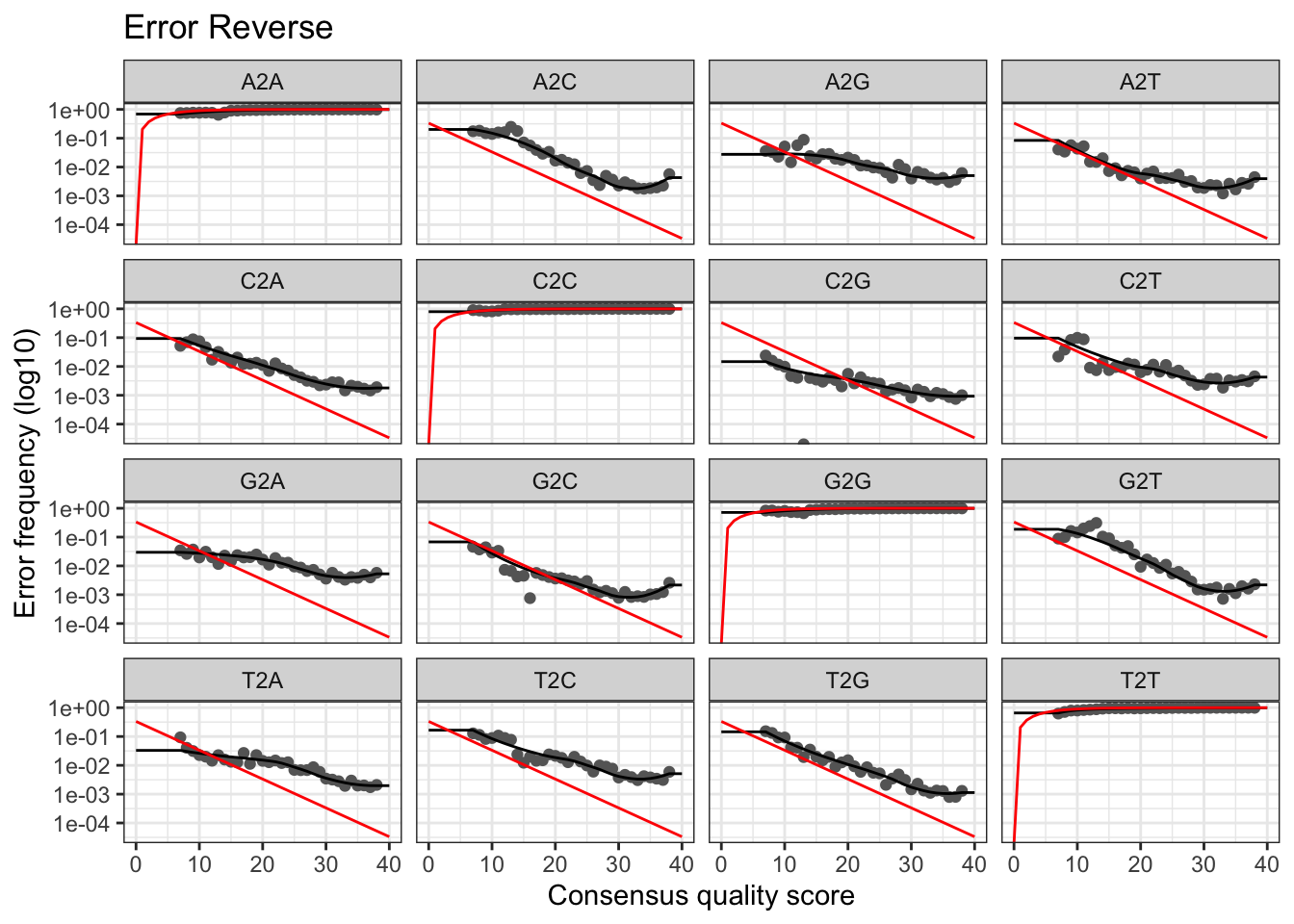
La ligne rouge correspond à ce qui est attendu sur la base du score de qualité, la ligne noire représente l’estimation et les points noirs représentent l’observation. D’une manière générale, vous voulez que l’observation (points noirs) corresponde bien à l’estimation (ligne noire).
Enregistrer les modèles d’erreur
Je vous conseil de sauvegarder les modèles d’erreur si vous avez de l’espace disque. Si vous avez besoin de relancer l’analyse en aval de cette étape, vous pouvez charger les modèles d’erreur et gagner du temps de calcul.
save(error_F, file = paste0(path_R, "error_F.RData"))
save(error_R, file = paste0(path_R, "error_R.RData"))
Déplication, inférences d’échantillons et fusion des séquences
Déplication
La déréplication est une étape courante dans de nombreux pipeline de traitement d’amplicon. Au lieu de conserver 100 séquences identiques et d’effectuer tout le traitement en aval sur les 100, vous pouvez conserver l’une d’entre elles et y attacher simplement le numéro 100. Lorsque DADA2 déreplique des séquences, il génère également un nouveau profil de score de qualité de chaque séquence unique basé sur les scores de qualité moyens de chaque base de toutes les séquences qui en étaient des répliques de celle-ci.
Inférences d’échantillons
C’est là que DADA2 arrive à faire ce pour quoi il est né, c’est-à-dire faire de son mieux pour déduire de véritables séquences biologiques. Pour ce faire, il incorpore les profils de qualité et les abondances de chaque séquence unique, puis détermine si chaque séquence est susceptible d’être d’origine biologique.
Fusion des séquences
Maintenant, DADA2 fusionne les ASV “forward” et “reverse” pour reconstruire notre amplicon cible complet.
Boucle R
Pour chaque échantillon, nous effectuons les trois étapes ci-dessus dans une boucle. Nous créons d’abord la liste qui contiendra les informations des séquences fusionnées, puis nous lançons la boucle qui déplique, infère et fusionne les séquences pour chaque échantillon.
merged_list <- vector("list", length(sample_names))
names(merged_list) <- sample_names
for(i in sample_names){
cat("Processing -------", which(sample_names == i), "/",
length(sample_names), "-------", i, "\n")
derep_Fs <- derepFastq(fas_Fs_filtered[[i]], verbose = TRUE)
derep_Rs <- derepFastq(fas_Rs_filtered[[i]], verbose = TRUE)
dds_Fs <- dada(derep_Fs,
err = error_F,
multithread = TRUE,
verbose = TRUE)
dds_Rs <- dada(derep_Rs,
err = error_R,
multithread = TRUE,
verbose = TRUE)
merged_list[[i]] <- mergePairs(dds_Fs,
derep_Fs,
dds_Rs,
derep_Rs,
verbose = TRUE)
}
Construire un tableau de séquence
Extraire l’abondance
Nous devons extraire l’abondance de la liste des séquences fusionnées que nous venons de créer. Nous n’utilisons ensuite que des abondances non nulles pour construire la table de séquence.
to.rm <- as.numeric(lapply(merged_list, function(x) sum(x$abundance)))
to.kp <- !(to.rm == 0)
merged_list_clean <- merged_list[to.kp]
Création d’une table de compte de séquences
Nous pouvons maintenant créer une table de compte de séquences avec la fonction makeSequenceTable(). C’est l’un des principaux résultats du traitement d’un ensemble de données d’amplicon.
seqtab <- makeSequenceTable(merged_list_clean)
Suppression des séquences chimères
DADA2 identifie les séquences chimères probables en alignant chaque séquence avec celles qui ont été récupérées en plus grande abondance, puis en voyant s’il existe des séquences à plus faible abondance qui peuvent être créées exactement en mélangeant les parties gauche et droite de deux des plus abondantes.
seqtab_nochim <- removeBimeraDenovo(seqtab,
method = "consensus",
multithread = TRUE,
verbose = TRUE)
Suivi des séquences au travers du pipeline
Nous créons un objet avec le nombre de lectures par échantillon après chaque étape du pipeline. Cela peut servir de point de départ pour vous aider à vous diriger vers l’étape du pipeline où vous devriez faire des modifications s’il vous reste peu de séquences.
reads.raw <- data.frame('sample' = rownames(seqtab),
'Merged' = rowSums(seqtab))
reads.nochim <- data.frame('sample' = rownames(seqtab_nochim),
'Nonchim' = rowSums(seqtab_nochim))
track <- data.frame('sample' = sample_names,
'Raw' = out_1[,1],
'Cutadapt' = out_1[,2],
'Filtered' = out_2[, 2])
track <- left_join(track, reads.raw) %>%
left_join(reads.nochim, by = 'sample')
track[is.na(track)] <- 0
track <- track[!(track$Nonchim > track$Raw),]
Attribuer une taxonomie
Charger la base de référence
load(paste0(path_R, "RDP_v18-mod_July2020.RData"))
# load(paste0(path_R, "SILVA_SSU_r138_2019.RData"))
Création de la liste de séquences uniques
dna <- DNAStringSet(getSequences(seqtab_nochim))
Assignation
ids <- IdTaxa(test=dna,
trainingSet=trainingSet,
strand="both",
threshold = 60,
processors=NULL,
verbose=TRUE)
Cleaning and saving files
À ce stade, nous effectuons une première sauvegarde des fichiers de données avant de passer à la décontamination.
Tableaux de compte des séquences
write.csv(prep_cdm(seqtab),
paste0(path_out, "sequence_table.csv"),
row.names = FALSE)
write.csv(prep_cdm(seqtab_nochim),
paste0(path_out, "sequence_table_nochim.csv"),
row.names = FALSE)
Séquences au travers du pipeline
track <- as.data.frame(track)
track <- cbind(rownames(track), track)
colnames(track)[1] <- "Sample"
write.csv(track,
paste0(path_out, "track_reads.csv"),
row.names = FALSE)
Fichier de séquences ASV
# donner à nos en-têtes seq des noms plus gérables (ASV_1, ASV_2...)
asv_seqs <- colnames(seqtab_nochim)
asv_headers <- vector(dim(seqtab_nochim)[2], mode="character")
for (i in 1:dim(seqtab_nochim)[2]) {
asv_headers[i] <- paste(">ASV", i, sep="_")
}
# faire et écrire un fasta de nos séquences ASV finales:
asv_fasta <- c(rbind(asv_headers, asv_seqs))
write(asv_fasta, paste0(path_out,"ASVs.fa"))
Taxonomie
# créer une table de taxonomie et identifier les séquences non classées "NA"
ranks <- c("domain", "phylum", "class", "order", "family", "genus", "species")
asv_tax <- t(sapply(ids, function(x) {
m <- match(ranks, x$rank)
taxa <- x$taxon[m]
taxa[startsWith(taxa, "unclassified_")] <- NA
taxa
}))
colnames(asv_tax) <- ranks
asv_tax = as.data.frame(asv_tax)
asv_tax = asv_tax[,-1]
ASV <- gsub(pattern=">",
replacement="",
x=asv_headers)
DNA_SEQ <- colnames(seqtab_nochim)
asv_tax <- cbind(DNA_SEQ,ASV,asv_tax)
dim(asv_tax)
[1] 400 8
dim(seqtab_nochim)
[1] 4 400
write.csv(asv_tax,
paste0(path_out, "seq_nochim_tax.csv"),
row.names = TRUE)
Éliminer les contaminants probables
Pour DADA2, @bejcal et al. ont également créé un programme pour éliminer les contaminants basé sur des échantillons blancs appelés decontam. Pour que decontam fonctionne sur nos données, nous devons lui fournir une table de compte des séquences, et nous devons également lui préciser un vecteur logique qui lui indique quels échantillons sont des blancs.
Spécifiez les blancs
Notez qu’ici j’utilise une simple boucle for pour spécifier quels échantillons sont des blancs dans le vecteur “vector_for_decontam”. S’il existe des annotations d’échantillons plus compliquées pour spécifier les blancs, cette partie devra être modifiée.
# préparation de la table de compte
asv_tab <- prep_cdm(seqtab_nochim)
rownames(asv_tab) <- seq(from=1, to=dim(asv_tab)[1],by=1)
#création du vecteur
vector_for_decontam <- c()
#Remplacez "BLANK" par l'identificateur de caractère de vos échantillons blancs
for (i in 1:dim(asv_tab[, 3:dim(asv_tab)[2]])[2]) {
vector_for_decontam[i] <- str_detect(colnames(asv_tab)[i],
"BLANK",
negate = FALSE)
}
Identification des contaminants
Ici, nous exécutons la fonction isContaninant qui renverra les ASV étant probablement des contaminants. La fréquence de chaque séquence en fonction de la concentration d’ADN amplifié dans chaque échantillon est utilisée pour identifier les séquences contaminantes.
contam_df <- isContaminant(t(asv_tab[, 3:dim(asv_tab)[2]]),
neg = vector_for_decontam)
table(contam_df$contaminant)
Taxonomie des contaminants probables
Ici, nous extrayons la taxonomie des contaminants probables.
contam_asvs <- row.names(contam_df[contam_df$contaminant == TRUE, ])
tax_contam <- asv_tax[contam_asvs, ]
write.csv(tax_contam,
paste0(path_out, "contam_seq_tax.csv"),
row.names = FALSE)
Élimination des contaminants probables
Maintenant qu’ils sont identifiés, nous pouvons créer des tableaux sans les contaminants. Nous créons d’abord un nouveau fichier fasta avec toutes les séquences ASV non contaminantes. Nous supprimons ensuite les ASV contaniants de nos tables de comptage et taxonomiques. Enfin nous vérifions les dimensions de ces deux tableaux qui doivent avoir le même nombre de lignes. S’ils ne le font pas, quelque chose s’est mal passé quelque part.
if (length(contam_asvs) == 0) {
asv_fasta_no_contam <- asv_fasta
} else {
contam_indices <- which(asv_fasta %in% paste0(">ASV_", contam_asvs))
dont_want <- sort(c(contam_indices, contam_indices + 1))
asv_fasta_no_contam <- asv_fasta[- dont_want]
}
asv_tab_no_contam <- asv_tab[!asv_tab$ASV %in% paste0("ASV_", contam_asvs), ]
asv_tax_no_contam <- asv_tax[!asv_tax$ASV %in% paste0("ASV_", contam_asvs), ]
dim(asv_tab_no_contam)
dim(asv_tax_no_contam)
Exportation des sorties
write(asv_fasta_no_contam,
paste0(path_out, "ASVs-no-contam.fa"))
write.csv(asv_tab_no_contam,
paste0(path_out, "ASVs_counts-no-contam.csv"),
row.names = FALSE)
write.csv(asv_tax_no_contam,
paste0(path_out, "ASVs_taxonomy-no-contam.csv"),
row.names = FALSE)
Clustering MOTUs
À ce stade, nous avons les tables ASV propres avec lesquelles travailler. Il peut être bon d’avoir une table OTU pour faire une analyse plus approfondie. Ceci peut être réalisé en regroupant les séquences ASV à une valeur seuil de similarité.
Notez que pour cette étape, certains packages sont requis mais non chargés:
- library(DECIPHER)
- library(Biostrings)
Spécifiez les séquences
Nous spécifions d’abord les séquences dans la préparation de l’ADN pour l’algorithme de clustering. Nous spécifions également le nombre de cœurs de processeur sur lesquels nous voulons que l’algorithme s’exécute.
nproc <- 4 # défini sur le nombre de processeurs à utiliser pour le clustering
asv_sequences <- asv_tab_no_contam$DNA_SEQ
dna <- Biostrings::DNAStringSet(asv_sequences)
Clustering ASVs
C’est là que la magie opère ! Nous alignons d’abord les séquences, puis créons une matrice de distance et enfin nous regroupons. Ici, nous utilisons un seuil de 0,03 pour avoir des OTU de similarité de 97%, cela peut être ajusté à vos besoins.
aln <- DECIPHER::AlignSeqs(dna, processors = nproc)
d <- DECIPHER::DistanceMatrix(aln, processors = nproc)
clusters <- DECIPHER::TreeLine(
myDistMatrix=d,
method = "complete",
cutoff = 0.03, # use `cutoff = 0.03` for a 97% OTU
type = "clusters",
processors = nproc)
Préparer et exporter la table OTU
clusters <- clusters %>%
add_column(DNA_SEQ = asv_sequences)
merged_seqtab <- asv_tab_no_contam[, -2] %>%
left_join(clusters) %>%
group_by(cluster) %>%
summarize_at(vars(-DNA_SEQ), sum) %>%
mutate(cluster = paste0("OTU", cluster))
write_csv(merged_seqtab, paste0(path_out, "OTU_table.csv"))