PARAFAC partie 1 : Conditionnement des données et indices de fluorescence
Jade Dormoy-Boulanger et Mathieu Michaud
Mai 2022
- Avant de commencer
- Importation des données fournies dans staRdom
- Vérification des données
- Préparation et correction des données
- Nom des échantillons
- Correction de l’absorbance de référence
- Correction spectrale
- Soustraction des blancs
- Correction de l’effet du filtre interne (IFE)
- Normalisation de Raman
- Retrait des blancs
- Retrait et interpolation de la dispersion
- Correction des dilutions
- Lissage des données
- Survol des données avant les indices de fluorescence
- Choix des pics de fluorescence et leurs indices
La méthode PARAFAC et les indices d’absorbance et de fluorescence permettent de caractériser la matière organique dissoute (MOD) selon sa composition, sa source et sa forme moléculaire. Ces méthodes utilisent différentes matrices d’émissions et d’excitation fournies par un spectrophotomètre à fluorescence. Elle est traditionnellement exécutée à l’aide du logiciel MATLAB, mais le paquet staRdom de R permet également de faire ce genre d’analyses.
Ce tutoriel est une adaptation du tutoriel de Matthias Pucher, qui a développé staRdom, intitulé “PARAFAC analysis of EEM data to separate DOM components in R”. Nous utiliserons les données d’exemple fournies dans le paquet staRdom, mais nous avons validé la méthode avec des données réelles également et nous sommes confiants de l’efficacité de la méthode.
Lien pour le tutoriel de Matthias Pucher: https://cran.r-project.org/web/packages/staRdom/vignettes/PARAFAC_analysis_of_EEM.html
La méthode dans R est équivalente à celle dans Matlab, comme le démontre cet article de Pucher et al. (2019): https://doi.org/10.3390/w11112366
Partie 1: Introduction aux indices d’absorbance et de fluorescence
Les analyses PARAFAC sont souvent accompagnées d’indices de fluorescence et d’absorbance. Comme ces analyses sont très lourdes, nous avons séparé les indices du PARAFAC en lui-même. Nous commencerons avec les indices, car le conditionnement des données requis est le même que pour le PARAFAC. Nous pourrons donc reprendre nos matrices d’émission et d’excitation (EEMs) pour la PARAFAC, à la suite de la génération des indices.
Avant de commencer
La première étape est d’installer staRdom et de vérifier le nombre de coeur de votre ordinateur. La meilleure optimisation de la vitesse des calculs est atteinte en indiquant le nombre de coeur de votre machine dans certaines fonctions plus demandantes de l’analyse. Nous utiliserons aussi les paquets dplyr, tidyr et eemR (eemR s’installe en même temps que staRdom).
#Chargement des paquets
library(staRdom)
library(dplyr)
library(tidyr)
library(eemR)
#optimisation de la vitesse de calcul
cores <-detectCores(logical = FALSE) # combien de coeur sur l'ordi
cores # j'ai 4 coeurs
## [1] 4
Importation des données fournies dans staRdom
Nous importerons les données exemples fournies dans staRdom. Si vous utilisez vos propres données, voici quelques recommandations:
- Les noms de fichers de EEMs et d’absorbance doivent être identiques
- Les noms des fichiers ne doivent pas contenir de “-“ ni commencer par un chiffre
- Les fichiers doivent être en csv et encodés avec des virgules comme séparateurs (pas de points virgules!)
- Les fichiers EEMs doivent aussi contenir les blancs et ils doivent absolument avoir dans leur noms soit “nano”, “miliq”, “milliq”, “mq” ou “blank”
- Utilisez directement les fonctions absorbance_read() et eem_read() en insérant le chemin menant à vos données (ex: eem_list <- eem_read(“C:/Users/Bureau/Doctorat /Donnees/PhD/DonneesR/PARAFAC-2020-brute/EEM”, recursive = T, import_function = “cary”)
- Pour eem_read(), vous pouvez indiquer la machine que vous utilisez avec l’argument import_function. staRdom supporte actuellement Varian Cary Eclipse (“cary”), Horiba Aqualog (“aqualog”), Horiba Fluoromax-4 (“fluoromax4”), Shimadzu (“shimadzu”), Hitachi F-7000 (eem_hitachi) et les csv générique (eem_csv)
system.file()
## [1] "C:/PROGRA~1/R/R-41~1.1/library/base"
#Absorbance
raw_abs<- system.file("extdata/absorbance", package = "staRdom")#chemin vers les données
absorbance <- absorbance_read(raw_abs, cores = cores)#les données, convertira les différents fichiers en un seul dataset.
#EEMs
eem<-system.file("extdata/EEMs", package = "staRdom")#chemin vers les données
eem_list <- eem_read(eem, recursive = TRUE, import_function = eem_csv) #les données,converties en un objet liste
eem_overview_plot(eem_list, spp=9, contour = TRUE)
## [[1]]
## Warning: Removed 104 rows containing non-finite values (stat_contour).

#Métadonnées
metatable <- system.file("extdata/metatable_dreem.csv",package = "staRdom") #chemin vers les métadonnées
meta <- read.table(metatable, header = TRUE, sep = ",", dec = ".", row.names = 1) # les métadonnées
#Si vous utilisez vos propre données, vous pouvez créer vos métadonnées avec le code suivant
eem_metatemplate(eem_list, absorbance) %>%
write.csv(file="metatable.csv", row.names = FALSE)
Vérification des données
La vérification des données peut se faire avec la fonction eem_checkdata():
- NAs (NAs_in_EEMs)
- Incompatibilités de gamme de longueurs d’onde (entre les EEMs: EEMs_more_data_than_smallest ; spectres d’absorbance vs EEMs: EEM_absorbance_wavelength_range_mismatch)
- Données incomplètes ( EEMs, mais pas d’absorbance: EEMs_missing_absorbance; absorbance, mais pas de EEMs: Absorbance_missing_EEMs; EEM mais pas de métadonnées: metadata: EEMs_missing_metadata; Métadonnées mais pas de EEMs: Metadata_missing_EEMs)
- Inconsistence des noms de fichiers (Duplicate_EEM_names, Duplicate_absorbance_names, invalid_EEM_names, invalid_absorbance_names,Duplicates_metatable_names)
- Méthode de correction non appliquée (missing_data_correction).
Les analyses peuvent soutenir 15%-20% de NAs. Au-delà, une interpolation est fortement suggérée
problem <- eem_checkdata(eem_list,absorbance,meta,metacolumns = c("dilution"),error=FALSE)
## NAs were found in the following samples (ratio):
## d423sf (0), d457sf (0), d492sf (0), d667sf (0), dblank_di25se06 (0), d433sf (0), d437sf (0), d441sf (0), dblank_mq11my (0.02),
## Please consider interpolating the data. It is highly recommended, due to more stable and meaningful PARAFAC models!
## EEM samples missing absorbance data:
## dblank_di25se06 in
## C:/Users/martich/Documents/R/win-library/4.1/staRdom/extdata/EEMs/di25se06
## dblank_mq11my in
## C:/Users/martich/Documents/R/win-library/4.1/staRdom/extdata/EEMs/mq11my
problem
## $Possible_problem_found
## [1] TRUE
##
## $NAs_in_EEMs
## d423sf d457sf d492sf d667sf dblank_di25se06
## 0.00000000 0.00000000 0.00000000 0.00000000 0.00000000
## d433sf d437sf d441sf dblank_mq11my
## 0.00000000 0.00000000 0.00000000 0.02173913
##
## $EEMs_more_data_than_smallest
## character(0)
##
## $missing_data_correction
## [1] NA
##
## $EEMs_missing_absorbance
## [1] "dblank_di25se06" "dblank_mq11my"
##
## $Absorbance_missing_EEMs
## character(0)
##
## $Duplicate_EEM_names
## character(0)
##
## $Duplicate_absorbance_names
## character(0)
##
## $invalid_EEM_names
## character(0)
##
## $invalid_absorbance_names
## character(0)
##
## $EEM_absorbance_wavelength_range_mismatch
## NULL
##
## $Duplicates_metatable_names
## character(0)
##
## $EEMs_missing_metadata
## NULL
##
## $Metadata_missing_EEMs
## NULL
Préparation et correction des données
Ici, nous aborderons plusieurs corrections importantes pour la production des indices de fluorescence et ultimement, l’analyse PARAFAC.
Nom des échantillons
Cette fonction permet de changer les noms des fichiers EEMs dans une liste de EEMs. Dans les données de staRdom, nous voulons enlever “(FD3)”, car il provient de la conversion de l’appareil en fichiers txt et que les données d’absorbance n’ont pas cette pas cette partie dans leur nom (On veut des noms identiques pour les EEMs et l’absorbance!!!)
eem_list <- eem_name_replace(eem_list,c("\\(FD3\\)"),c(""))
Correction de l’absorbance de référence
Normalement, la correction de l’absorbance est entre 680 et 700 nm.
absorbance <- abs_blcor(absorbance,wlrange = c(680,700))
Correction spectrale
La correction spectrale sert à enlever l’influence spécifique de l’instrument sur les EEMs. Normalement les instruments vont également fournir deux fichiers ( un émission et un excitation) de correction avec les analyses. Si vous utilisez vos données, vous pouvez simplement entrer le chemin dans la fonction (ex: data.table::fread(“C:/Bureau/Doctorat/Donnees/PhD/DonneesR/PARAFAC_Numerilab/CorrFiles/Ex_Corr.csv”)
# Excitation
excorfile <- system.file("extdata/CorrectionFiles/xc06se06n.csv",package="staRdom")# le chemin
Excor <- data.table::fread(excorfile)#les données
#Émission
emcorfile <- system.file("extdata/CorrectionFiles/mcorrs_4nm.csv",package="staRdom")#le chemin
Emcor <- data.table::fread(emcorfile)#les données
#Ajustement de la portée des EEMs pour couvrir la correction des vecteurs
eem_list <- eem_range(eem_list,ex = range(Excor[,1]), em = range(Emcor[,1]))#création de la portée spectrale
eem_list <- eem_spectral_cor(eem_list,Excor,Emcor)#correction
Soustraction des blancs
Les blancs doivent absolument avoir dans leur noms soit “nano”, “miliq”, “milliq”, “mq” ou “blank”. Ils sont utilisés lors de la normalisation de Raman. Si plusieurs blancs sont utilisés, une moyenne sera faite. Les blancs seront également soustraits de chaque échantillons pour réduire les effets des bandes de dispersion et des erreurs systématiques
# extension et interpolation
eem_list <- eem_extend2largest(eem_list, interpolation = 1, extend = FALSE, cores = cores)
# Soustraction des blancs
eem_list <- eem_remove_blank(eem_list)
## A total of 1 blank EEMs will be averaged.
## A total of 1 blank EEMs will be averaged.
#visualisation de la procédure
eem_overview_plot(eem_list, spp=9, contour = TRUE)
## [[1]]
## Warning: Removed 416 rows containing non-finite values (stat_contour).
Correction de l’effet du filtre interne (IFE)
Le IFE apparait quand la lumière d’excitation est absorbée par les chromophores (particules colorées). La méthode pour corriger l’IFE dans les EEMs utilise simplement l’absorbance.
eem_list <- eem_ife_correction(eem_list,absorbance, cuvl = 5)#culv = longueur de cuvette d'instrument (cm)
## d423sf
## Range of IFE correction factors: 1.0004 1.0288
## Range of total absorbance (Atotal) : 1e-04 0.0049
##
## d457sf
## Range of IFE correction factors: 1.0003 1.0208
## Range of total absorbance (Atotal) : 1e-04 0.0036
##
## d492sf
## Range of IFE correction factors: 1.0004 1.0241
## Range of total absorbance (Atotal) : 1e-04 0.0041
##
## d667sf
## Range of IFE correction factors: 1.0004 1.0249
## Range of total absorbance (Atotal) : 1e-04 0.0043
## Warning in FUN(X[[i]], ...): No absorbance data was found for sample
## dblank_di25se06!No absorption data was found for sample dblank_di25se06!
## d433sf
## Range of IFE correction factors: 1.0003 1.0241
## Range of total absorbance (Atotal) : 1e-04 0.0041
##
## d437sf
## Range of IFE correction factors: 1.0002 1.0131
## Range of total absorbance (Atotal) : 0 0.0023
##
## d441sf
## Range of IFE correction factors: 1.0002 1.016
## Range of total absorbance (Atotal) : 0 0.0028
## Warning in FUN(X[[i]], ...): No absorbance data was found for sample
## dblank_mq11my!No absorption data was found for sample dblank_mq11my!
#C'est normal qu'il n'y aie pas d'absorbance pour les blancs...qui seront retirés plus tard
eem_overview_plot(eem_list, spp=9, contour = TRUE)#visualisation
## [[1]]
## Warning: Removed 416 rows containing non-finite values (stat_contour).
Normalisation de Raman
L’intensité de fluorescence peut changer d’un instrument à l’autre, d’un paramètre à l’autre ou encore d’un jour à l’autre. Elle doit donc être normalisée à une échelle standard d’unité de Raman en divisant toutes les intensités par l’aire du pic de Raman(excitation de 350nm comprise dans une émission entre 371nm et 428nm) d’un échantillon d’eau ultrapure. Ici nous utiliserons les blancs d’eau ultrapure.
eem_list <- eem_raman_normalisation2(eem_list, blank = "blank")
#correction de raman, blank = méthode de correction (ici avec les blancs)
## A total of 1 blank EEMs will be averaged.
## Raman area: 5687348
## Raman area: 5687348
## Raman area: 5687348
## Raman area: 5687348
## A total of 1 blank EEMs will be averaged.
## Raman area: 5688862
## Raman area: 5688862
## Raman area: 5688862
eem_overview_plot(eem_list, spp=9, contour = TRUE) #visualisation
## [[1]]
## Warning: Removed 416 rows containing non-finite values (stat_contour).
Retrait des blancs
À partir de maintenant, nous n’avons plus besoin des blancs, donc nous les retirons du jeu de données.
#retrait des EEMs
eem_list <- eem_extract(eem_list, c("nano", "miliq", "milliq", "mq", "blank"),ignore_case = TRUE)
## Removed sample(s): dblank_di25se06 dblank_mq11my
#retrait des absorbances
absorbance <- dplyr::select(absorbance, -matches("nano|miliq|milliq|mq|blank", ignore.case = TRUE))
Retrait et interpolation de la dispersion
Suivant le retrait de la dispersion, même si ce n’est pas nécessaire, les aires de dispersion devraient être interpolées. Les résultats du PARAFAC seront plus juste et les calculs plus rapides. eem_interp() offre plusieurs méthodes d’interpolation qui dépendent de différentes fonctions intégrées:
- type = 0, tout les NAs seront remplacés par 0
- type = 1, méthode prévilégiée, spline d’interpolation
- type = 2, interpolation à l’aide des longueurs d’ondes d’émission et d’excitation et de leur moyennes subsequentes
- type = 3, interpolation à l’aide des longueurs d’ondes d’excitation
- type = 4, interpolation linéaire à l’aide des longueurs d’ondes d’émission et d’excitation et de leur moyennes subsequentes
Si la méthode d’interpolation type = 1 donne des résultats un peu bizarre dans les graphiques, alors pensez à utiliser un autre type.
#retrait de la dispersion
#création du vecteur où on indique que les valeurs à retirer sont bien dans l'ordre suivant:
# “raman1”, “raman2”, “rayleigh1” and “rayleigh2”
remove_scatter <- c(TRUE, TRUE, TRUE, TRUE)
#création d'un vecteur indiquant la largeur en nm de chaque longueur d'onde à retirer.
# L'ordre est le même que celui du précédent vecteur
remove_scatter_width <- c(15,15,15,15)
#le retrait
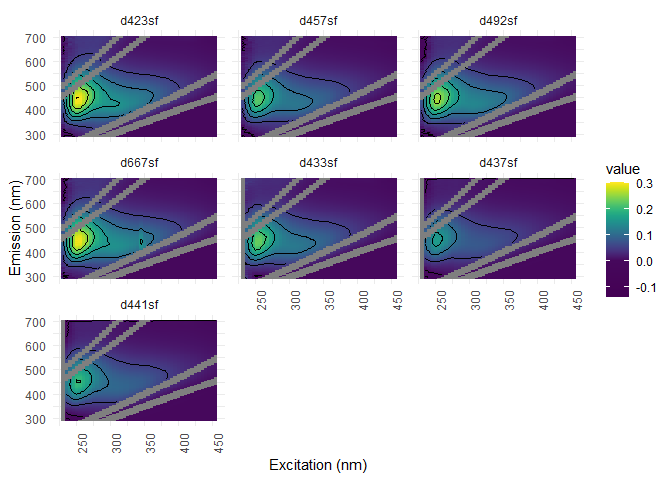
eem_list <- eem_rem_scat(eem_list, remove_scatter = remove_scatter, remove_scatter_width = remove_scatter_width)
#la visualisation
eem_overview_plot(eem_list, spp=9, contour = TRUE)
## [[1]]
## Warning: Removed 6294 rows containing non-finite values (stat_contour).
#Interpolation
eem_list <- eem_interp(eem_list, cores = cores, type = 1, extend = FALSE)
eem_overview_plot(eem_list, spp=9, contour = TRUE)#visualisation
## [[1]]
## Warning: Removed 312 rows containing non-finite values (stat_contour).
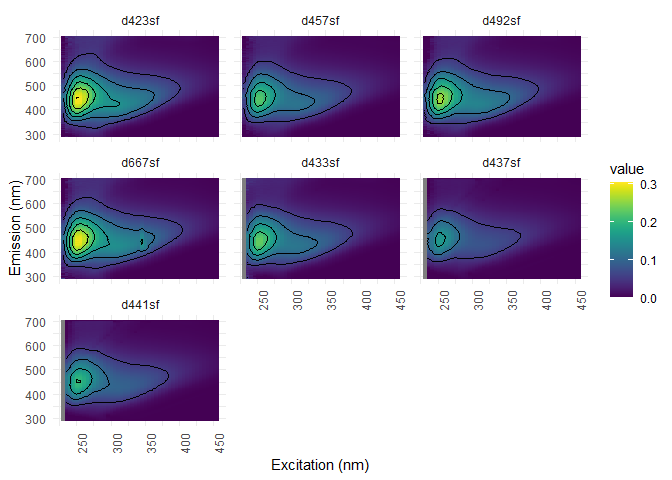
Correction des dilutions
Les données doivent être corrigées avec leur facteur de dilution respectif s’il y a lieu
dil_data <- meta["dilution"]# création du dataset contenant les facteurs de dilution
eem_list <- eem_dilution(eem_list,dil_data)#application de la correction
eem_overview_plot(eem_list, dil_data)#visualisation
## [[1]]
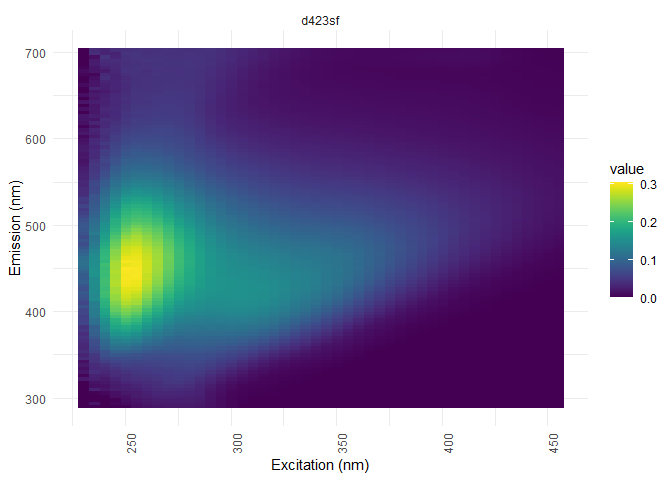
##
## [[2]]
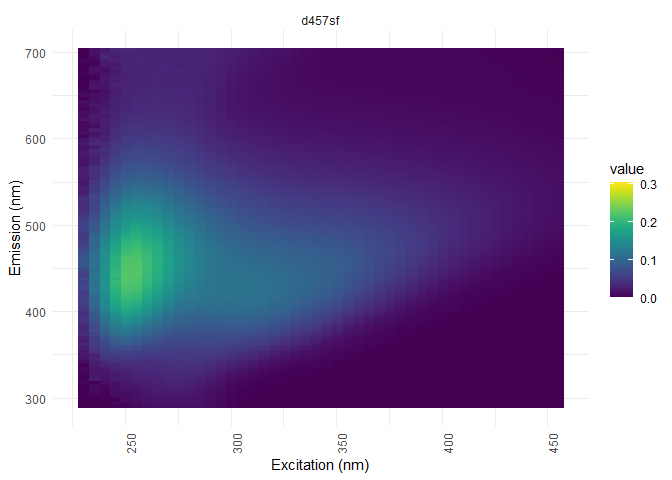
##
## [[3]]
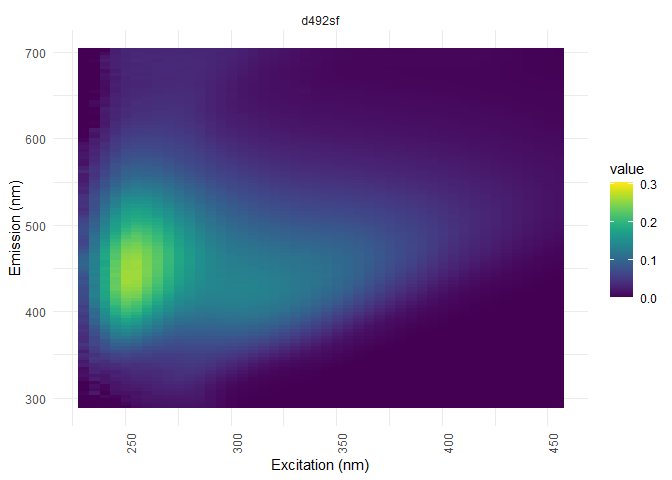
##
## [[4]]
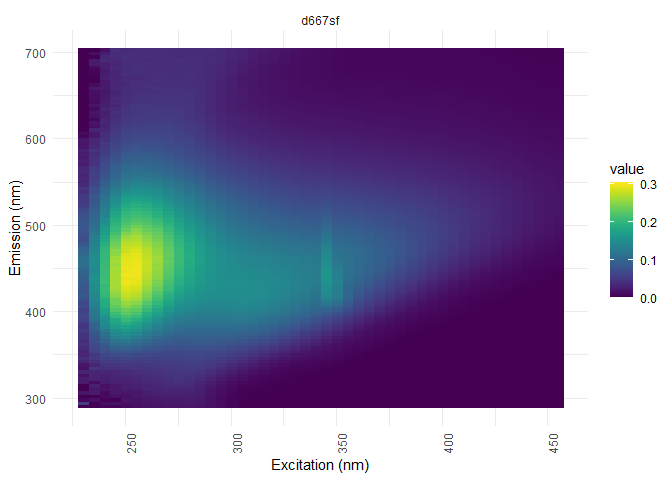
##
## [[5]]
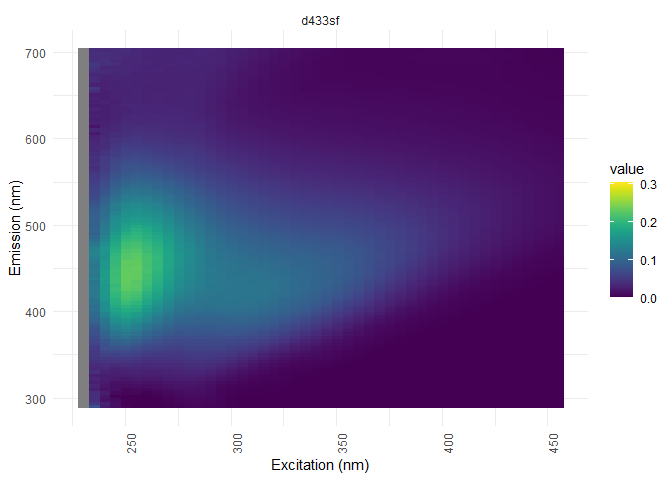
##
## [[6]]
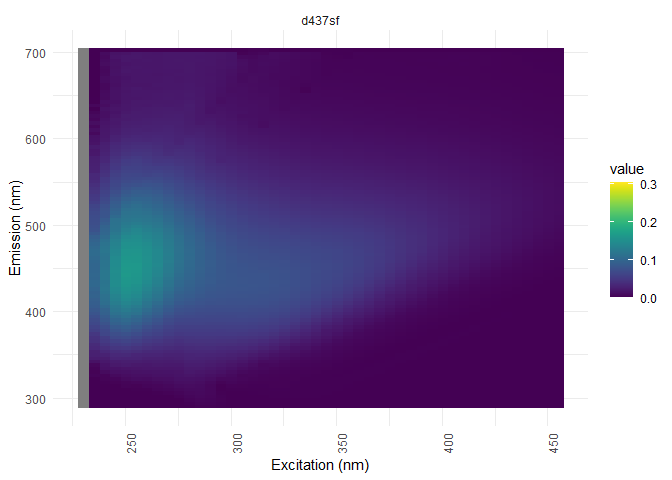
##
## [[7]]
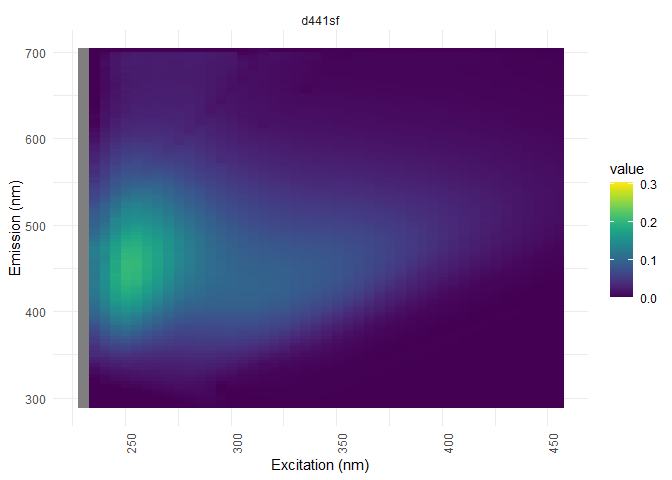
Lissage des données
Dépendamment de l’instrument utilisé pour analyser les données, le lissage des données peut être utile pour le choix des pics de fluorescence. Par contre, ce n’est pas conseillé pour les analyses PARAFAC. La fonction utilise un “roulement des moyennes” et dans l’exemple ci-dessous, le roulement sera fait à coup de quatre nm à la fois sur les données de fluorescence.
eem4peaks <- eem_smooth(eem_list, n = 4, cores = cores)#n spécifie la taille moyenne mobile de la fenêtre en nm
Survol des données avant les indices de fluorescence
Nous donne un aperçu du conditionnement que l’on vient de faire sur les données
summary(eem_list)
## sample ex_min ex_max em_min em_max is_blank_corrected is_scatter_corrected
## 1 d423sf 230 455 290 702 TRUE TRUE
## 2 d457sf 230 455 290 702 TRUE TRUE
## 3 d492sf 230 455 290 702 TRUE TRUE
## 4 d667sf 230 455 290 702 TRUE TRUE
## 5 d433sf 230 455 290 702 TRUE TRUE
## 6 d437sf 230 455 290 702 TRUE TRUE
## 7 d441sf 230 455 290 702 TRUE TRUE
## is_ife_corrected is_raman_normalized
## 1 TRUE TRUE
## 2 TRUE TRUE
## 3 TRUE TRUE
## 4 TRUE TRUE
## 5 TRUE TRUE
## 6 TRUE TRUE
## 7 TRUE TRUE
Choix des pics de fluorescence et leurs indices
Les données lissées sont utilisées ici
#Biological index, renseigne sur la production autochtone/ la MOD d'orgine
#aquatique microbienne;
#0.6-0.7 = MOD plus dégradée d'origine terrestre
#et >1 = MOD fraîchement produite par les bactéries aquatiques
bix <- eem_biological_index(eem4peaks)
#Pic de Coble,
#B = protéine-like tyrosine ou tryptophane (origine microbienne),
#T = protéine-like tryptophane (origine microbienne),
#A = substance humique (origine plante vasculaire),
#C = substance humique (origine plante vasculaire) et
#M = substance humique (origine production autochtone)
coble_peaks <- eem_coble_peaks(eem4peaks)
#Fluorescence index, renseigne sur la source de la MOD;
#1.7-2 = de source microbienne,
#1.2-1.5 = origine des sols et plantes terrestres
fi <- eem_fluorescence_index(eem4peaks)
#Humification index, renseigne sur le degré d'humification;
#10-16 = origine terrestre,
#<4 = origine autochtone
hix <- eem_humification_index(eem4peaks, scale = TRUE)
#création de la table
indices_peaks <- bix %>%
full_join(coble_peaks, by = "sample") %>%
full_join(fi, by = "sample") %>%
full_join(hix, by = "sample")
#la table
indices_peaks
## sample bix b t a m c
## 1 d423sf 0.7238682 0.036238767 0.06222814 0.2799546 0.14974696 0.11645331
## 2 d457sf 0.6858719 0.023536584 0.04106616 0.2082118 0.11265412 0.08778000
## 3 d492sf 0.6869648 0.027140701 0.04730339 0.2413028 0.13198615 0.10493114
## 4 d667sf 0.6839838 0.031426888 0.05391093 0.2774084 0.14513535 0.13263827
## 5 d433sf 0.6941625 0.012110049 0.03792344 0.2147849 0.11547600 0.09000859
## 6 d437sf 0.6678838 0.006024978 0.02159146 0.1516322 0.07649198 0.06366574
## 7 d441sf 0.6670705 0.007355762 0.02692251 0.1882532 0.09387812 0.07938853
## fi hix
## 1 1.151716 0.8805637
## 2 1.143778 0.8923698
## 3 1.161794 0.8949828
## 4 1.139740 0.8965758
## 5 1.155606 0.9143584
## 6 1.116053 0.9420593
## 7 1.108152 0.9395073
Indices d’absorbance
Différents indices d’absorbances peuvent données différentes information quant à la provenance de la MOD. Voici ceux qui nous intéressent:
- a254: coefficient d’aborption du CDOM
- SR: slope ratio; s’il est haut, le poids moléculaire est faible et vice-versa
Pour calculer l’indice de teneur en composés aromatiques SUVA~254~ : a254/COD (mg/l)
slope_parms <- abs_parms(absorbance, cuvl = 1, cores = cores) #calcul des indices
## Warning: `funs()` was deprecated in dplyr 0.8.0.
## Please use a list of either functions or lambdas:
##
## # Simple named list:
## list(mean = mean, median = median)
##
## # Auto named with `tibble::lst()`:
## tibble::lst(mean, median)
##
## # Using lambdas
## list(~ mean(., trim = .2), ~ median(., na.rm = TRUE))
## This warning is displayed once every 8 hours.
## Call `lifecycle::last_lifecycle_warnings()` to see where this warning was generated.
slope_parms #les indices
## sample a254 a300 E2_E3 E4_E6 S275_295 S350_400 S300_700
## 1 d423sf 13.869423 6.341582 7.275041 54.45985 0.01693705 0.01767518 0.01757271
## 2 d433sf 11.629678 5.354673 7.195549 67.97273 0.01685673 0.01775750 0.01764950
## 3 d437sf 6.323142 2.836798 7.235020 39.38501 0.01750176 0.01674770 0.01719949
## 4 d441sf 7.703282 3.396527 7.572209 39.06406 0.01776943 0.01723729 0.01747484
## 5 d457sf 10.051932 4.654212 7.091301 71.26347 0.01675176 0.01752695 0.01741157
## 6 d492sf 11.652366 5.424564 7.060283 73.15475 0.01665879 0.01754663 0.01743985
## 7 d667sf 12.048233 5.542739 7.063998 35.38728 0.01648855 0.01797665 0.01702009
## SR
## 1 0.9582393
## 2 0.9492738
## 3 1.0450242
## 4 1.0308717
## 5 0.9557717
## 6 0.9494014
## 7 0.9172203