Manipulation rapide de bases de données avec dplyr et ses alliés
Charles Martin
Octobre 2018
- Notre jeu de données
- Les 5 opérations de base
- Nettoyage des données
- Importation de données
- Dernier exercice
Notre jeu de données
library(ggplot2)
data(msleep)
msleep
# A tibble: 83 x 11
name genus vore order conservation sleep_total sleep_rem sleep_cycle
<chr> <chr> <chr> <chr> <chr> <dbl> <dbl> <dbl>
1 Cheet… Acin… carni Carn… lc 12.1 NA NA
2 Owl m… Aotus omni Prim… <NA> 17 1.8 NA
3 Mount… Aplo… herbi Rode… nt 14.4 2.4 NA
4 Great… Blar… omni Sori… lc 14.9 2.3 0.133
5 Cow Bos herbi Arti… domesticated 4 0.7 0.667
6 Three… Brad… herbi Pilo… <NA> 14.4 2.2 0.767
7 North… Call… carni Carn… vu 8.7 1.4 0.383
8 Vespe… Calo… <NA> Rode… <NA> 7 NA NA
9 Dog Canis carni Carn… domesticated 10.1 2.9 0.333
10 Roe d… Capr… herbi Arti… lc 3 NA NA
# ... with 73 more rows, and 3 more variables: awake <dbl>, brainwt <dbl>,
# bodywt <dbl>
Les 5 opérations de base
Comme lors de la formation précédente, toutes ces opérations pourraient s’effectuer avec les fonctions R de base, mais de façon beaucoup moins lisible et intégrée (nous y reviendront)
Filtrer
P. ex., disons que nous désirons conserver uniquement les observations pour les mammifères de plus de 200 g
library(dplyr)
Attaching package: 'dplyr'
The following objects are masked from 'package:stats':
filter, lag
The following objects are masked from 'package:base':
intersect, setdiff, setequal, union
filter(msleep, bodywt > 0.2)
# A tibble: 61 x 11
name genus vore order conservation sleep_total sleep_rem sleep_cycle
<chr> <chr> <chr> <chr> <chr> <dbl> <dbl> <dbl>
1 Cheet… Acin… carni Carn… lc 12.1 NA NA
2 Owl m… Aotus omni Prim… <NA> 17 1.8 NA
3 Mount… Aplo… herbi Rode… nt 14.4 2.4 NA
4 Cow Bos herbi Arti… domesticated 4 0.7 0.667
5 Three… Brad… herbi Pilo… <NA> 14.4 2.2 0.767
6 North… Call… carni Carn… vu 8.7 1.4 0.383
7 Dog Canis carni Carn… domesticated 10.1 2.9 0.333
8 Roe d… Capr… herbi Arti… lc 3 NA NA
9 Goat Capri herbi Arti… lc 5.3 0.6 NA
10 Guine… Cavis herbi Rode… domesticated 9.4 0.8 0.217
# ... with 51 more rows, and 3 more variables: awake <dbl>, brainwt <dbl>,
# bodywt <dbl>
Attention, comme toutes les manipulations d’objets dans R, le résultat est “perdu” si il n’est pas assigné à un nouvel objet…
grands_mams <- filter(msleep, bodywt > 0.2)
Dans RStudio, on peut aussi consulter le résultat d’une opération, avec ou sans la sauvegarde dans un objet :
View(filter(msleep, bodywt > 0.2))
View(grands_mams)
Quatre pièges à éviter
#1 Les nombres à virgule ne sont pas indéfiniment précis
1/49*49 == 1
[1] FALSE
R conserve dans ses calculs un nombre limité de décimales, ce qui fait que certaines erreurs d’arondissement peuvent compliquer les comparaisons.
On peut contourner le problème avec l’opérateur near
near(1/49*49, 1)
[1] TRUE
*#2 Le = ne veut pas dire *égal **
Le symbole = dans R est utilisé pour les assignations, comme un synonyme de <-.
Pour effectuer des comparaisons (comme dans la majorité des langages informatiques),
il faut utiliser ==.
filter(msleep, vore = "omni")
Error: `vore` (`vore = "omni"`) must not be named, do you need `==`?
**#3 Les données manquantes présentent certaines particularités **
On ne peut pas vérifier si une valeur est manquante avec ==
NA == NA
[1] NA
La logique de R sous-jacente étant que, si je ne connaîs pas l’âge de Paul et que je ne connais pas l’âge de Jacques, la réponse à “Est-ce que Paul et Jacques ont le même âge” est “Je ne sais pas”, et non “vrai”
Si on veut tester des valeurs manquantes, il faut utiliser la fonction is.na
filter(msleep, is.na(conservation))
# A tibble: 29 x 11
name genus vore order conservation sleep_total sleep_rem sleep_cycle
<chr> <chr> <chr> <chr> <chr> <dbl> <dbl> <dbl>
1 Owl m… Aotus omni Prim… <NA> 17 1.8 NA
2 Three… Brad… herbi Pilo… <NA> 14.4 2.2 0.767
3 Vespe… Calo… <NA> Rode… <NA> 7 NA NA
4 Afric… Cric… omni Rode… <NA> 8.3 2 NA
5 Weste… Euta… herbi Rode… <NA> 14.9 NA NA
6 Galago Gala… omni Prim… <NA> 9.8 1.1 0.55
7 Human Homo omni Prim… <NA> 8 1.9 1.5
8 Macaq… Maca… omni Prim… <NA> 10.1 1.2 0.75
9 "Vole… Micr… herbi Rode… <NA> 12.8 NA NA
10 Littl… Myot… inse… Chir… <NA> 19.9 2 0.2
# ... with 19 more rows, and 3 more variables: awake <dbl>, brainwt <dbl>,
# bodywt <dbl>
**#4 Il faut modifier notre pensée pour combiner certaines conditions **
R possède un opérateur permettant de faire des OU (|) et un permettant de faire
des ET (&). Leur usage est cependant différent du français.
Si on veut p. ex. tous les mammifères omnivores ou carnivores, on serait tentés de faire :
filter(msleep, vore == "omni" | "carni")
Error in filter_impl(.data, quo): Evaluation error: operations are possible only for numeric, logical or complex types.
Mais il faut en fait spécifier toutes les éventualités, et les séparer par l’opérateur |. P. ex. :
filter(msleep, vore == "omni" | vore == "carni")
# A tibble: 39 x 11
name genus vore order conservation sleep_total sleep_rem sleep_cycle
<chr> <chr> <chr> <chr> <chr> <dbl> <dbl> <dbl>
1 Cheet… Acin… carni Carn… lc 12.1 NA NA
2 Owl m… Aotus omni Prim… <NA> 17 1.8 NA
3 Great… Blar… omni Sori… lc 14.9 2.3 0.133
4 North… Call… carni Carn… vu 8.7 1.4 0.383
5 Dog Canis carni Carn… domesticated 10.1 2.9 0.333
6 Grivet Cerc… omni Prim… lc 10 0.7 NA
7 Star-… Cond… omni Sori… lc 10.3 2.2 NA
8 Afric… Cric… omni Rode… <NA> 8.3 2 NA
9 Lesse… Cryp… omni Sori… lc 9.1 1.4 0.15
10 Long-… Dasy… carni Cing… lc 17.4 3.1 0.383
# ... with 29 more rows, and 3 more variables: awake <dbl>, brainwt <dbl>,
# bodywt <dbl>
On peut cependant raccourcir ce genre d’affirmation avec l’opérateur %in%
filter(msleep, vore %in% c("omni", "carni"))
# A tibble: 39 x 11
name genus vore order conservation sleep_total sleep_rem sleep_cycle
<chr> <chr> <chr> <chr> <chr> <dbl> <dbl> <dbl>
1 Cheet… Acin… carni Carn… lc 12.1 NA NA
2 Owl m… Aotus omni Prim… <NA> 17 1.8 NA
3 Great… Blar… omni Sori… lc 14.9 2.3 0.133
4 North… Call… carni Carn… vu 8.7 1.4 0.383
5 Dog Canis carni Carn… domesticated 10.1 2.9 0.333
6 Grivet Cerc… omni Prim… lc 10 0.7 NA
7 Star-… Cond… omni Sori… lc 10.3 2.2 NA
8 Afric… Cric… omni Rode… <NA> 8.3 2 NA
9 Lesse… Cryp… omni Sori… lc 9.1 1.4 0.15
10 Long-… Dasy… carni Cing… lc 17.4 3.1 0.383
# ... with 29 more rows, and 3 more variables: awake <dbl>, brainwt <dbl>,
# bodywt <dbl>
Avec l’opérateur %in%, on peut même préparer notre liste de valeurs à l’avance
a_garder <- c("omni", "carni", "herbi")
filter(msleep, vore %in% a_garder)
# A tibble: 71 x 11
name genus vore order conservation sleep_total sleep_rem sleep_cycle
<chr> <chr> <chr> <chr> <chr> <dbl> <dbl> <dbl>
1 Cheet… Acin… carni Carn… lc 12.1 NA NA
2 Owl m… Aotus omni Prim… <NA> 17 1.8 NA
3 Mount… Aplo… herbi Rode… nt 14.4 2.4 NA
4 Great… Blar… omni Sori… lc 14.9 2.3 0.133
5 Cow Bos herbi Arti… domesticated 4 0.7 0.667
6 Three… Brad… herbi Pilo… <NA> 14.4 2.2 0.767
7 North… Call… carni Carn… vu 8.7 1.4 0.383
8 Dog Canis carni Carn… domesticated 10.1 2.9 0.333
9 Roe d… Capr… herbi Arti… lc 3 NA NA
10 Goat Capri herbi Arti… lc 5.3 0.6 NA
# ... with 61 more rows, and 3 more variables: awake <dbl>, brainwt <dbl>,
# bodywt <dbl>
On peut aussi inverser des conditions
Avec le point d’exclamation, p. ex. pour avoir tout sauf les omnivores :
filter(msleep, !(vore == "omni"))
# A tibble: 56 x 11
name genus vore order conservation sleep_total sleep_rem sleep_cycle
<chr> <chr> <chr> <chr> <chr> <dbl> <dbl> <dbl>
1 Cheet… Acin… carni Carn… lc 12.1 NA NA
2 Mount… Aplo… herbi Rode… nt 14.4 2.4 NA
3 Cow Bos herbi Arti… domesticated 4 0.7 0.667
4 Three… Brad… herbi Pilo… <NA> 14.4 2.2 0.767
5 North… Call… carni Carn… vu 8.7 1.4 0.383
6 Dog Canis carni Carn… domesticated 10.1 2.9 0.333
7 Roe d… Capr… herbi Arti… lc 3 NA NA
8 Goat Capri herbi Arti… lc 5.3 0.6 NA
9 Guine… Cavis herbi Rode… domesticated 9.4 0.8 0.217
10 Chinc… Chin… herbi Rode… domesticated 12.5 1.5 0.117
# ... with 46 more rows, and 3 more variables: awake <dbl>, brainwt <dbl>,
# bodywt <dbl>
Maintenant que toutes ces particularités sont derrière nous, on peut passer à la seconde opération sur les bases de données, le tri.
Trier
Par défaut, le tri se fait en ordre croissant
arrange(msleep, bodywt)
# A tibble: 83 x 11
name genus vore order conservation sleep_total sleep_rem sleep_cycle
<chr> <chr> <chr> <chr> <chr> <dbl> <dbl> <dbl>
1 Lesse… Cryp… omni Sori… lc 9.1 1.4 0.15
2 Littl… Myot… inse… Chir… <NA> 19.9 2 0.2
3 Great… Blar… omni Sori… lc 14.9 2.3 0.133
4 Deer … Pero… <NA> Rode… <NA> 11.5 NA NA
5 House… Mus herbi Rode… nt 12.5 1.4 0.183
6 Big b… Epte… inse… Chir… lc 19.7 3.9 0.117
7 North… Onyc… carni Rode… lc 14.5 NA NA
8 "Vole… Micr… herbi Rode… <NA> 12.8 NA NA
9 Afric… Rhab… omni Rode… <NA> 8.7 NA NA
10 Vespe… Calo… <NA> Rode… <NA> 7 NA NA
# ... with 73 more rows, and 3 more variables: awake <dbl>, brainwt <dbl>,
# bodywt <dbl>
Il faut utiliser une modificateur pour inverser l’ordre
arrange(msleep, desc(bodywt))
# A tibble: 83 x 11
name genus vore order conservation sleep_total sleep_rem sleep_cycle
<chr> <chr> <chr> <chr> <chr> <dbl> <dbl> <dbl>
1 Afric… Loxo… herbi Prob… vu 3.3 NA NA
2 Asian… Elep… herbi Prob… en 3.9 NA NA
3 Giraf… Gira… herbi Arti… cd 1.9 0.4 NA
4 Pilot… Glob… carni Ceta… cd 2.7 0.1 NA
5 Cow Bos herbi Arti… domesticated 4 0.7 0.667
6 Horse Equus herbi Peri… domesticated 2.9 0.6 1
7 Brazi… Tapi… herbi Peri… vu 4.4 1 0.9
8 Donkey Equus herbi Peri… domesticated 3.1 0.4 NA
9 Bottl… Turs… carni Ceta… <NA> 5.2 NA NA
10 Tiger Pant… carni Carn… en 15.8 NA NA
# ... with 73 more rows, and 3 more variables: awake <dbl>, brainwt <dbl>,
# bodywt <dbl>
Premier exercice
Trouver en ordre croissant de poids du corps, la liste de tous les herbivores non domestiqués
Sélectionner certaines colonnes
select(msleep, vore, brainwt, bodywt)
# A tibble: 83 x 3
vore brainwt bodywt
<chr> <dbl> <dbl>
1 carni NA 50
2 omni 0.0155 0.48
3 herbi NA 1.35
4 omni 0.00029 0.019
5 herbi 0.423 600
6 herbi NA 3.85
7 carni NA 20.5
8 <NA> NA 0.045
9 carni 0.07 14
10 herbi 0.0982 14.8
# ... with 73 more rows
Une série de colonnes en séquence
select(msleep, sleep_total:awake)
# A tibble: 83 x 4
sleep_total sleep_rem sleep_cycle awake
<dbl> <dbl> <dbl> <dbl>
1 12.1 NA NA 11.9
2 17 1.8 NA 7
3 14.4 2.4 NA 9.6
4 14.9 2.3 0.133 9.1
5 4 0.7 0.667 20
6 14.4 2.2 0.767 9.6
7 8.7 1.4 0.383 15.3
8 7 NA NA 17
9 10.1 2.9 0.333 13.9
10 3 NA NA 21
# ... with 73 more rows
ou tout sauf les colonnes de la séquence
select(msleep, -c(sleep_total:awake))
# A tibble: 83 x 7
name genus vore order conservation brainwt bodywt
<chr> <chr> <chr> <chr> <chr> <dbl> <dbl>
1 Cheetah Acinonyx carni Carnivo… lc NA 50
2 Owl monkey Aotus omni Primates <NA> 0.0155 0.48
3 Mountain beaver Aplodon… herbi Rodentia nt NA 1.35
4 Greater short-ta… Blarina omni Soricom… lc 0.00029 0.019
5 Cow Bos herbi Artioda… domesticated 0.423 600
6 Three-toed sloth Bradypus herbi Pilosa <NA> NA 3.85
7 Northern fur seal Callorh… carni Carnivo… vu NA 20.5
8 Vesper mouse Calomys <NA> Rodentia <NA> NA 0.045
9 Dog Canis carni Carnivo… domesticated 0.07 14
10 Roe deer Capreol… herbi Artioda… lc 0.0982 14.8
# ... with 73 more rows
On peut aussi renommer certaines colonnes
rename(msleep, nom = name, genre = genus)
# A tibble: 83 x 11
nom genre vore order conservation sleep_total sleep_rem sleep_cycle
<chr> <chr> <chr> <chr> <chr> <dbl> <dbl> <dbl>
1 Cheet… Acin… carni Carn… lc 12.1 NA NA
2 Owl m… Aotus omni Prim… <NA> 17 1.8 NA
3 Mount… Aplo… herbi Rode… nt 14.4 2.4 NA
4 Great… Blar… omni Sori… lc 14.9 2.3 0.133
5 Cow Bos herbi Arti… domesticated 4 0.7 0.667
6 Three… Brad… herbi Pilo… <NA> 14.4 2.2 0.767
7 North… Call… carni Carn… vu 8.7 1.4 0.383
8 Vespe… Calo… <NA> Rode… <NA> 7 NA NA
9 Dog Canis carni Carn… domesticated 10.1 2.9 0.333
10 Roe d… Capr… herbi Arti… lc 3 NA NA
# ... with 73 more rows, and 3 more variables: awake <dbl>, brainwt <dbl>,
# bodywt <dbl>
Ajouter des colonnes
Puisque les colonnes sont ajoutées à la fin du jeu de données, nous allons créer un jeu de données simplifié pour bien voir ce que l’on fait
poids <- select(msleep,ends_with("wt"))
poids
# A tibble: 83 x 2
brainwt bodywt
<dbl> <dbl>
1 NA 50
2 0.0155 0.48
3 NA 1.35
4 0.00029 0.019
5 0.423 600
6 NA 3.85
7 NA 20.5
8 NA 0.045
9 0.07 14
10 0.0982 14.8
# ... with 73 more rows
Pour ajouter une colonne du poids du cerveau en grammes
mutate(poids, cerveau_g = brainwt*1000)
# A tibble: 83 x 3
brainwt bodywt cerveau_g
<dbl> <dbl> <dbl>
1 NA 50 NA
2 0.0155 0.48 15.5
3 NA 1.35 NA
4 0.00029 0.019 0.290
5 0.423 600 423
6 NA 3.85 NA
7 NA 20.5 NA
8 NA 0.045 NA
9 0.07 14 70
10 0.0982 14.8 98.2
# ... with 73 more rows
On peut aussi utiliser plusieurs colonnes à la fois dans un calcul, p. ex. pour calculer la taille relative du cerveau :
mutate(poids, rel_brain = brainwt / bodywt)
# A tibble: 83 x 3
brainwt bodywt rel_brain
<dbl> <dbl> <dbl>
1 NA 50 NA
2 0.0155 0.48 0.0323
3 NA 1.35 NA
4 0.00029 0.019 0.0153
5 0.423 600 0.000705
6 NA 3.85 NA
7 NA 20.5 NA
8 NA 0.045 NA
9 0.07 14 0.005
10 0.0982 14.8 0.00664
# ... with 73 more rows
Combiner plusieurs opérations dans une chaîne
C’est ici que l’approche dplyr se démarque que l’approche classique!
Déjà, on peut faire beaucoup de choses avec les opérations vues précédemment, p. ex. obtenir le nom et le poids des 10 plus petits mammifères:
x <- arrange(msleep,bodywt) # trier du plus petit au plus grand
y <- mutate(x,rang = row_number())# ajouter une colonne de rang
z <- filter(y, rang <= 10)# garder les 10 plus petits
select(z,name,bodywt)# ne garder que les noms et les poids
# A tibble: 10 x 2
name bodywt
<chr> <dbl>
1 Lesser short-tailed shrew 0.005
2 Little brown bat 0.01
3 Greater short-tailed shrew 0.019
4 Deer mouse 0.021
5 House mouse 0.022
6 Big brown bat 0.023
7 Northern grasshopper mouse 0.028
8 "Vole " 0.035
9 African striped mouse 0.044
10 Vesper mouse 0.045
Ça nous fait beaucoup d’objets intermédiaires inutiles. On pourrait bêtement les éliminer
select(filter(mutate(arrange(msleep,bodywt),rang = row_number()), rang <= 10),name,bodywt)
# A tibble: 10 x 2
name bodywt
<chr> <dbl>
1 Lesser short-tailed shrew 0.005
2 Little brown bat 0.01
3 Greater short-tailed shrew 0.019
4 Deer mouse 0.021
5 House mouse 0.022
6 Big brown bat 0.023
7 Northern grasshopper mouse 0.028
8 "Vole " 0.035
9 African striped mouse 0.044
10 Vesper mouse 0.045
Mais ce faisant, on perd grandement en lisibilité. On pourrait indenter le code pour y voir plus clair :
select(
filter(
mutate(
arrange(msleep,bodywt),
rang = row_number()
),
rang <= 10
),
name,
bodywt
)
# A tibble: 10 x 2
name bodywt
<chr> <dbl>
1 Lesser short-tailed shrew 0.005
2 Little brown bat 0.01
3 Greater short-tailed shrew 0.019
4 Deer mouse 0.021
5 House mouse 0.022
6 Big brown bat 0.023
7 Northern grasshopper mouse 0.028
8 "Vole " 0.035
9 African striped mouse 0.044
10 Vesper mouse 0.045
Mais on reste avec le problème que ce n’est pas simple au premier coup d’oeil de voir sur quel tableau de données on travaille. De plus, il faut lire du centre vers l’extérieur, ce qui est peu intuitif.
La solution, l’opérateur d’enchaînement (Pipe Operator), de la librairie magrittr : %>%
msleep %>%
arrange(bodywt) %>%
mutate(rang = row_number()) %>%
filter(rang <= 10) %>%
select(name, bodywt)
# A tibble: 10 x 2
name bodywt
<chr> <dbl>
1 Lesser short-tailed shrew 0.005
2 Little brown bat 0.01
3 Greater short-tailed shrew 0.019
4 Deer mouse 0.021
5 House mouse 0.022
6 Big brown bat 0.023
7 Northern grasshopper mouse 0.028
8 "Vole " 0.035
9 African striped mouse 0.044
10 Vesper mouse 0.045
%>% transforme notre version lisible en une version que l’ordinateur est prêt à exécuter…
- Plus facile à lire
- On récupère la lecture de haut en bas et de gauche à droite
- On n’a plus d’objets intermédiaires inutiles
- L’accent est mis sur la transformation (le verbe)
- On trouve rapidement le point de départ (le tableau de données)
Raccourci clavier : Ctrl+Shift+M
Toutes les librairies faisant partie du tidyverse de Hadley Wickham supportent l’opérateur d’enchaînement, sauf ggplot2. Ce dernier peut néanmoins être ajouté au bout d’une chaîne :
msleep %>%
filter(bodywt > 0.200) %>%
mutate(
l_corps = log(bodywt),
l_cerveau = log(brainwt)
) %>%
ggplot(aes(x = l_corps,y = l_cerveau, col = vore)) +
geom_point()
Warning: Removed 19 rows containing missing values (geom_point).
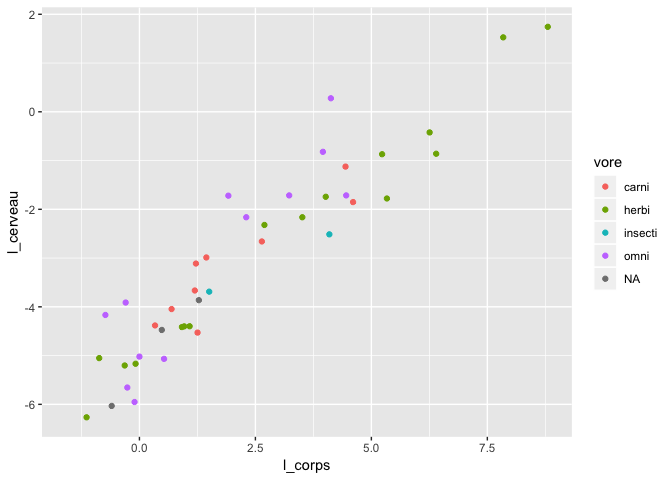
Il faut être super attentif, parce que toutes les autres fonctions s’enchaînent
avec %>%, sauf les couches de ggplot qui s’attachent avec des +
Deuxième exercice
On refait le premier exercice, mais cette fois-ci, en prenant avantage de l’opérateur d’enchaînement :
Trouver en ordre croissant de poids du corps, la liste de tous les herbivores non domestiqués
Résumer les données
Dernière opération de base que l’on peut faire avec un jeu de données propres : le résumer.
On peut résumer plusieurs variables ou fonctions à la fois :
msleep %>%
summarize(
poids_moyen = mean(bodywt),
ecart_type_poids = sd(bodywt)
)
# A tibble: 1 x 2
poids_moyen ecart_type_poids
<dbl> <dbl>
1 166. 787.
Cette opération devient beaucoup plus puissante si on utilise les regroupements :
msleep %>%
group_by(vore) %>%
summarize(
poids_moyen = mean(bodywt),
ecart_type_poids = sd(bodywt)
)
# A tibble: 5 x 3
vore poids_moyen ecart_type_poids
<chr> <dbl> <dbl>
1 carni 90.8 182.
2 herbi 367. 1244.
3 insecti 12.9 26.4
4 omni 12.7 24.7
5 <NA> 0.858 1.34
Nettoyage des données
Format long vs. format large
Parfois, le format dans lequel nous entrons nos données est très pratique lors de la saisie, mais ne correspond pas à la définition des données propres (une ligne par observations, une colonne par variable).
Souvent, on se retrouve avec des données qui ressemblent à ceci :
oiseaux <- tibble(
Especes = c("Corneille", "Mésange"),
"2001" = c(0,1),
"2002" = c(2,1),
"2003" = c(2,2)
)
oiseaux
# A tibble: 2 x 4
Especes `2001` `2002` `2003`
<chr> <dbl> <dbl> <dbl>
1 Corneille 0 2 2
2 Mésange 1 1 2
Comment tracerait-on le graphique de l’abondance des corneilles au fil des années?
La solution est de passer les données au format long plutôt que large :
library(tidyr)
o2 <-
oiseaux %>%
gather(
key = Annee,
value = Abondance,
`2001`:`2003`,
convert = TRUE
)
o2
# A tibble: 6 x 3
Especes Annee Abondance
<chr> <int> <dbl>
1 Corneille 2001 0
2 Mésange 2001 1
3 Corneille 2002 2
4 Mésange 2002 1
5 Corneille 2003 2
6 Mésange 2003 2
L’argument key correspond au nom de la colonne qui contiendra les anciens noms de colonnes.
L’argument valuecorrespond au nom de la colonne qui contiendra les valeurs dans l’ancien tableau.
Autrement dit, les deux questions à vous poser dans le passage du format large au format long sont :
- à quoi correspondent vraiment les noms de colonnes
- à quoi correspondent vraiment les valeurs dans les cellules
Ensuite, on peut finalement tracer le graphique…
o2 %>%
filter(Especes == "Corneille") %>%
ggplot(aes(x = Annee, y = Abondance)) +
geom_line()
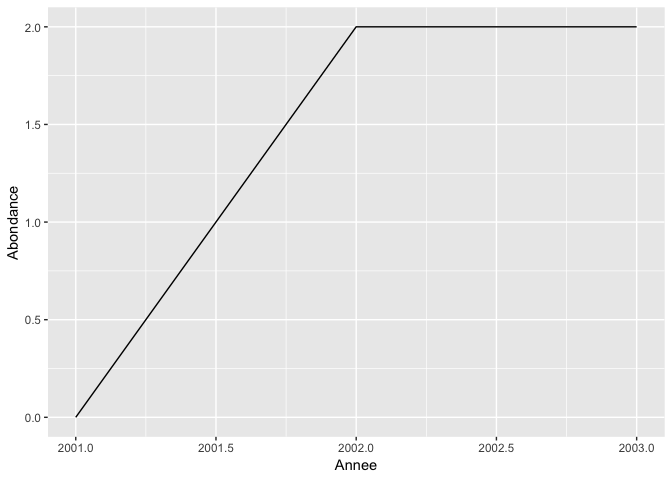
Une autre application du format long : répéter une opération sur plusieur variables.
P. ex. pour obtenir l’histogramme de toutes les variables de sommeil dans le jeu
de données msleep :
msleep_long <- msleep %>%
gather(
key = Variable,
value = Valeur,
sleep_total:awake
)
msleep_long
# A tibble: 332 x 9
name genus vore order conservation brainwt bodywt Variable Valeur
<chr> <chr> <chr> <chr> <chr> <dbl> <dbl> <chr> <dbl>
1 Cheetah Acin… carni Carn… lc NA 50 sleep_t… 12.1
2 Owl mo… Aotus omni Prim… <NA> 0.0155 0.48 sleep_t… 17
3 Mounta… Aplo… herbi Rode… nt NA 1.35 sleep_t… 14.4
4 Greate… Blar… omni Sori… lc 0.00029 0.019 sleep_t… 14.9
5 Cow Bos herbi Arti… domesticated 0.423 600 sleep_t… 4
6 Three-… Brad… herbi Pilo… <NA> NA 3.85 sleep_t… 14.4
7 Northe… Call… carni Carn… vu NA 20.5 sleep_t… 8.7
8 Vesper… Calo… <NA> Rode… <NA> NA 0.045 sleep_t… 7
9 Dog Canis carni Carn… domesticated 0.07 14 sleep_t… 10.1
10 Roe de… Capr… herbi Arti… lc 0.0982 14.8 sleep_t… 3
# ... with 322 more rows
msleep_long %>%
ggplot(aes(x = Valeur)) +
geom_histogram() +
facet_wrap(~Variable)
`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.
Warning: Removed 73 rows containing non-finite values (stat_bin).
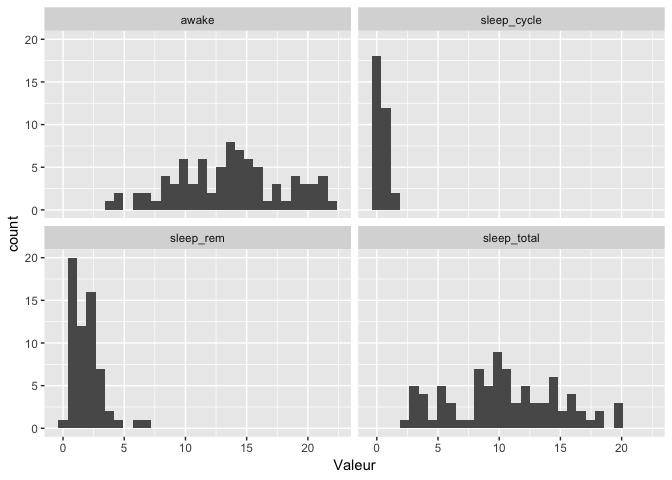
Si à l’inverse, une même observation a été divisée sur plusieurs lignes, on peut la rassembler :
mesures <- data.frame(
individu = c("A","A","B","B","C","C"),
mesure = c("pH","O2", "pH","O2","pH","O2"),
valeur = c(6,99,7,90, 6.5, 89)
)
mesures
individu mesure valeur
1 A pH 6.0
2 A O2 99.0
3 B pH 7.0
4 B O2 90.0
5 C pH 6.5
6 C O2 89.0
mesures %>%
spread(
key = mesure,
value = valeur
)
individu O2 pH
1 A 99 6.0
2 B 90 7.0
3 C 89 6.5
Et on est de retour à une variable par colonne et une obsevation par ligne
Pour la fonction spread, l’argument key correspond à la colonne contenant les
noms des colonnes à créer, et l’argument value correspond à la colonne contenant
les valeurs
VOIR SLIDES
Connecter deux bases de données ensemble
Pour l’exercice, nous allons faire comme si la base de données msleep nous avait été fournie en deux morceaux séparées. Un premier avec toute la méta-information, et l’autre avec les données de sommeil comme tel. Les deux tableaux ne sont pas dans le même ordre, et dans un, les carnivores étaient absents.
meta <- msleep %>%
select(name:conservation) %>%
arrange(name)
sommeil <- msleep %>%
filter(vore != "carni") %>%
select(name, sleep_total:awake)
meta
# A tibble: 83 x 5
name genus vore order conservation
<chr> <chr> <chr> <chr> <chr>
1 African elephant Loxodonta herbi Proboscidea vu
2 African giant pouched rat Cricetomys omni Rodentia <NA>
3 African striped mouse Rhabdomys omni Rodentia <NA>
4 Arctic fox Vulpes carni Carnivora <NA>
5 Arctic ground squirrel Spermophilus herbi Rodentia lc
6 Asian elephant Elephas herbi Proboscidea en
7 Baboon Papio omni Primates <NA>
8 Big brown bat Eptesicus insecti Chiroptera lc
9 Bottle-nosed dolphin Tursiops carni Cetacea <NA>
10 Brazilian tapir Tapirus herbi Perissodac… vu
# ... with 73 more rows
sommeil
# A tibble: 57 x 5
name sleep_total sleep_rem sleep_cycle awake
<chr> <dbl> <dbl> <dbl> <dbl>
1 Owl monkey 17 1.8 NA 7
2 Mountain beaver 14.4 2.4 NA 9.6
3 Greater short-tailed shrew 14.9 2.3 0.133 9.1
4 Cow 4 0.7 0.667 20
5 Three-toed sloth 14.4 2.2 0.767 9.6
6 Roe deer 3 NA NA 21
7 Goat 5.3 0.6 NA 18.7
8 Guinea pig 9.4 0.8 0.217 14.6
9 Grivet 10 0.7 NA 14
10 Chinchilla 12.5 1.5 0.117 11.5
# ... with 47 more rows
Comment faire un diagramme à moustache du temps de sommeil par statut de conservation (en faisant comme si msleep n’existait pas! )?
On ne peut pas simplement coller nos deux tableaux de données (bind_cols) parcequ’ils n’ont pas nécéssairement les mêmes lignes, le même ordre, etc. Il
faut plutôt utiliser les fonction de la famille *_join du package tidyr.
VOIR SLIDES
Dans le cas qui nous intéresse, quelle est la clé unique connectant nos deux tableaux?
Dans notre cas, nous avons plusieurs possiblités pour connecter les deux tableaux, p. ex.
sommeil %>%
left_join(meta)
Joining, by = "name"
# A tibble: 57 x 9
name sleep_total sleep_rem sleep_cycle awake genus vore order
<chr> <dbl> <dbl> <dbl> <dbl> <chr> <chr> <chr>
1 Owl monkey 17 1.8 NA 7 Aotus omni Prima…
2 Mountain … 14.4 2.4 NA 9.6 Aplodo… herbi Roden…
3 Greater s… 14.9 2.3 0.133 9.1 Blarina omni Soric…
4 Cow 4 0.7 0.667 20 Bos herbi Artio…
5 Three-toe… 14.4 2.2 0.767 9.6 Bradyp… herbi Pilosa
6 Roe deer 3 NA NA 21 Capreo… herbi Artio…
7 Goat 5.3 0.6 NA 18.7 Capri herbi Artio…
8 Guinea pig 9.4 0.8 0.217 14.6 Cavis herbi Roden…
9 Grivet 10 0.7 NA 14 Cercop… omni Prima…
10 Chinchilla 12.5 1.5 0.117 11.5 Chinch… herbi Roden…
# ... with 47 more rows, and 1 more variable: conservation <chr>
Autrement dit, d’ajouter les meta-informations à notre base de données sommeil.
Mais on aurait pu aussi faire l’inverse :
meta %>%
left_join(sommeil)
Joining, by = "name"
# A tibble: 83 x 9
name genus vore order conservation sleep_total sleep_rem sleep_cycle
<chr> <chr> <chr> <chr> <chr> <dbl> <dbl> <dbl>
1 Afric… Loxo… herbi Prob… vu 3.3 NA NA
2 Afric… Cric… omni Rode… <NA> 8.3 2 NA
3 Afric… Rhab… omni Rode… <NA> 8.7 NA NA
4 Arcti… Vulp… carni Carn… <NA> NA NA NA
5 Arcti… Sper… herbi Rode… lc 16.6 NA NA
6 Asian… Elep… herbi Prob… en 3.9 NA NA
7 Baboon Papio omni Prim… <NA> 9.4 1 0.667
8 Big b… Epte… inse… Chir… lc 19.7 3.9 0.117
9 Bottl… Turs… carni Ceta… <NA> NA NA NA
10 Brazi… Tapi… herbi Peri… vu 4.4 1 0.9
# ... with 73 more rows, and 1 more variable: awake <dbl>
Dans ce cas, notre base de données est plus complète, mais contient des valeurs manquantes pour le sommeil de certains animaux
Importation de données
Au Québec, l’importation de données dans R à partir de fichiers CSV est particulièrement complexe :
VOIR SLIDES
- Notre séparateur de décimales est la virgule plutôt de que le point
- Notre séparateur de milliers est l’espace plutôt que la virgule
- Nos dates ne sont pas dans le même ordre que les anglophones
- Toutes sortes d’ennuis avec les lettres accentuées.
- La version française de Excel nous dit créer des fichiers CSV, alors qu’en fait, elle insère des points-virgules plutôt que des virgules.
La méthode à (presque) toute épreuve que je vous propose : charger vos données directement à partir de votre fichier Excel!
La raison : le représentation interne des données dans le fichier Excel est super constante et ne varie pas entre les langues ou les pays.
Les règles à suivre pour que les données s’importent bien à partir de votre fichier Excel :
- Les méta-informations ne doivent pas être dans la même feuille que les données
- Au pire, les méta-information doivent être en haut de la feuille
- Assurez-vous que vos noms de colonnes ne sont pas dédoublés
- Pas de fusion de cellules ou de lignes vides inutiles : données propres
- Assurez-vous que Excel interprète correctement vos données (p. ex. que vous êtes capables de calculer une moyenne sur votre colonne)
library(readxl)
x <- read_excel("Exemple.xlsx")
x
# A tibble: 4 x 5
Colonne1 Colonne2 Colonne3 Colonne4 Colonne5
<chr> <chr> <dbl> <dbl> <dttm>
1 A 4,3 4.12 4.12 2018-10-01 00:00:00
2 A 3,2 4.12 4.12 2018-01-10 00:00:00
3 B 1,1 4.12 4.12 2018-10-01 00:00:00
4 B 2,1 4.12 4.12 2018-11-01 00:00:00
On peut aussi passer des lignes ou aller lire une feuille spécifique plutôt que la première :
obs <- read_excel("Exemple.xlsx",sheet = 2, skip = 2)
Dernier exercice
Comment convertir le tableau obs dans un format approprié aux analyses de
communautés, avec une ligne par site, et une colonne par espèce?
e.g. pour arriver à ceci :
# A tibble: 3 x 3
Site Corneille `Geai bleu`
<dbl> <dbl> <dbl>
1 1 1 2
2 2 2 5
3 3 1 0
Ces exercices ont été préparées avec…
sessionInfo()
R version 3.5.1 (2018-07-02)
Platform: x86_64-apple-darwin15.6.0 (64-bit)
Running under: macOS High Sierra 10.13.6
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRblas.0.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/3.5/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] readxl_1.1.0 tidyr_0.8.1 bindrcpp_0.2.2 dplyr_0.7.7
[5] ggplot2_3.0.0
loaded via a namespace (and not attached):
[1] Rcpp_0.12.17 pillar_1.2.3 compiler_3.5.1 cellranger_1.1.0
[5] plyr_1.8.4 bindr_0.1.1 tools_3.5.1 digest_0.6.15
[9] evaluate_0.10.1 tibble_1.4.2 gtable_0.2.0 pkgconfig_2.0.1
[13] rlang_0.2.1 cli_1.0.0 yaml_2.1.19 withr_2.1.2
[17] stringr_1.3.1 knitr_1.20 rprojroot_1.3-2 grid_3.5.1
[21] tidyselect_0.2.4 glue_1.2.0 R6_2.2.2 rmarkdown_1.10
[25] purrr_0.2.5 magrittr_1.5 backports_1.1.2 scales_0.5.0
[29] htmltools_0.3.6 assertthat_0.2.0 colorspace_1.3-2 labeling_0.3
[33] utf8_1.1.4 stringi_1.2.3 lazyeval_0.2.1 munsell_0.5.0
[37] crayon_1.3.4