brms : Une fonction pour les contrôler tous
Comment une seule fonction, et un peu de réflexion, peut remplacer tout votre bestiaire de tests statistiques
par Charles Martin
Avril 2026
- Introduction
brms- Préparation de l’environnement
- La régression linéaire
- Test de t à deux échantillons
- Test de F
- ANOVA
- Le test de chi-carré
- Vers l’infini et plus loin encore
- Pour aller plus loin?
Introduction
Peut-être avez-vous déjà remarqué que certaines choses que l’on vous a enseignées comme des tests statistiques, par exemple comparer deux moyennes, pouvaient aussi être calculées à l’aide d’une régression linéaire, du moment que l’on connaissait certains “petits trucs”, par exemple comment transformer des variables catégoriques en variables numériques. Dans le même ordre d’idées, peut-être que vous avez déjà entendu parler du fait que l’ensemble des tests statistiques de base pouvaient être remplacés par la régression linéaire.
Avez-vous déjà essayé? Avez-vous trébuché en chemin? N’ayez crainte, cet atelier
a pour but de vous guider dans le processus. Pour remplacer votre bestiaire de
tests statistiques par une seule fonction, pouvant être adaptée à beaucoup,
mais alors beaucoup de situations : la fonction brm de la librairie brms.
brms
Même si techniquement tout ce que nous verrons aujourd’hui provient de la grande
famille de la régression (ou plus précisement du modèle linéaire), certaines des
opérations demandent des fonctions avec des noms différents dans R. Le but de l’atelier
étant de vous montrer une simplification, il aurait été un peu improductif de
vous forcer à utiliser 3 ou 4 fonctions différentes pour accomplir le travail.
C’est pourquoi nous utiliserons la fonction brm de la librairie brms, conçue
explicitement pour accomoder un grand nombre de situations.
Une des différences majeures entre la librairie brms et ses plus proches rivales
(p. ex. lme4, glmmTMB, mgcv) est que cette dernière ajuste les modèles dans un cadre
bayésien plutôt qu’un cadre fréquentiste. Vous verrez cependant que dans les faits,
cette contrainte est plus libératrice que contraignante, au prix d’un léger délai
(souvent quelques secondes) comparé aux approches fréquentistes.
L’idée générale derrière les modèles bayésiens est que la recherche des meilleures valeurs pour les paramètres s’effectue non pas par une opération d’algèbre matricielle (vs. les moindres carrés) ou par un algorithme se rapprochant progressivement de la solution (vs. la maximisation de la vraisemblance) mais plutôt par un échantillonnage de l’espace possible des paramètres. Cet échantillonnage, basé sur les principes de la méthode de Monte-Carlo et des chaînes de Markov ira explorer plus souvent les valeurs les plus probables des paramètres, et moins souvent les valeurs moins probables.
L’avantage majeur de cette approche est que l’on a toujours accès directement à toute la distribution des valeurs que pourraient prendre un paramètre. Dans l’approche fréquentiste, la technique suppose certaines choses à propos des données, et produit un intervalle de confiance basé sur ces assomptions, mais la distribution réelle pourrait être totalement différente et nous n’en saurions rien. C’est pourquoi la surveillance des assomptions ou des conditions d’application des techniques fréquentistes est si critique.
Au contraire, avec l’approche bayésienne, nous avons toujours accès à la distribution de chacun des paramètres. La question devient donc : considérant ce modèle, voici les distributions de chacun des paramètres. Il faut évidemment explorer les propriétés du modèle et tenter de l’améliorer si nécessaire / désiré, mais les chiffres représentent fidèlement ce que le modèle et nos données nous permettent d’inférer.
Le deuxième avantage majeur de l’approche bayésienne est qu’il incorpore de l’information connue a priori au calcul. Plutôt que de simplement se fier aux nouvelles données, le processus en est un de “mise à jour de nos croyances”.
Par exemple, un résultat comme remporter 10 paris consécutifs pourrait être interprété d’un point de vue fréquentiste comme suggérant un “talent” du parieur. Basé sur ces 10 paris, on calculerait quelque chose comme un intervalle de confiance entre 69 et 100% d’être profitable. Par contre, une analyse bayésienne pourrait démarrer d’un a priori conservateur qui dit, par exemple, que seuls 3% des parieurs sont profitables sur le long terme. À ce moment, une séquence de 10 gains ferait passer notre confiance en notre profitabilité plutôt autour de 10%.
Ceci étant dit, il n’est pas obligatoire de toujours utiliser les a priori informatifs.
Par défaut, si on ne lui dit rien, brms utilise des a priori plats, non-informatifs pour
les coefficients de nos modèles. Afin de limiter l’exploration à des valeurs raisonnables,
les a priori des termes d’erreurs et de l’ordonnée à l’origine sont cependant de type
faiblement informatif. Dans la majorité des cas, les résultats seront presque
identiques aux estimations fréquentistes. Par exemple, la pente évaluée pour la
régression ci-bas est identique au centième près à l’estimation fournie par
la fonction lm
Préparation de l’environnement
Nous aurons donc besoin pour travailler des librairies tidyverse et palmerpenguins qui nous fourniront nos données et les outils pour les manipuler.
Nous activerons aussi la librairie brms et tidybayes une librairie pour faciliter la
conversion des sorties d’un processus MCMC dans un cadre tidy.
library(tidyverse)
library(palmerpenguins)
library(brms)
library(tidybayes)
La régression linéaire
Comme tout l’atelier repose sur le fait que l’on peut remplacer l’ensemble des tests standards par des régressions linéaires, commençons donc par cette première technique, qui ne nécessitera essentiellement pas de conversion.
Ici, notre objectif sera de savoir si plus les manchots ont de longues ailes, plus ils sont lourds (i.e. peut-être parce qu’ils s’alimentent plus facilement,etc.)?
Donc, nous préparerons un petit tableau de données propres permettant de répondre à cette question :
pour_regression <-
penguins |>
select(body_mass_g, flipper_length_mm) |>
drop_na()
Afin de faciliter la compréhension des diverses conversions, nous prendrons quelques minutes à chaque test pour regarder la formule mathématique definissant la modélisation.
Pour la régression linéaire, vous vous rappelez peut-être que la formule classique est habituellement décrite comme ceci :
\[\begin{gathered} y_i = B_0 + B_1 x_i + \epsilon_i \\ \epsilon_i \sim \mathcal{N}(0,\sigma^2) \end{gathered}\]Par contre, de plus en plus de manuels utilisent une différente façon d’écrire le même concept mathématique, parce qu’elle fait plus facilement la séparation entre la partie déterministe du modèle (l’équation de régression) et la partie stochastique (la distribution des erreurs) :
\[\begin{gathered} y_i \sim \mathcal{N}(\mu,\sigma^2)\\ \mu=B_0 + B_1x_i \end{gathered}\]Dans cette formulation, on voit aussi clairement apparaître les trois paramètres que le modèle devra estimer, soit l’ordonnée à l’origine, la pente, mais aussi sigma, l’écart-type des erreurs.
Et ensuite, nous calculerons les paramètres de cette régression comme tel, mais avec
la fonction brm plutôt que lm. Si votre ordinateur le permet, vous pouvez comme
je le fais ici séparer le processus MCMC sur plusieurs coeurs, afin qu’il soit
fait en parallèle pour accélérer le traitement.
m_regression <-
brm(
body_mass_g ~ flipper_length_mm,
data = pour_regression,
cores = 4
)
Pour bien comprendre ce qui s’est passé durant le processus d’ajustement, il est souvent intéressant d’aller voir nous même ce qui a été échantilloné par le processus MCMC. Chacune des combinaisons de paramètres testées est conservée dans notre objet de modèle, et on peut accéder à leurs valeurs comme ceci :
m_regression |>
as.data.frame() |>
head()
b_Intercept b_flipper_length_mm sigma Intercept lprior lp__
1 -5449.433 47.94143 392.7860 4182.729 -15.03088 -2538.551
2 -6017.252 50.94069 400.5896 4217.506 -15.04448 -2538.194
3 -6085.078 51.28134 426.8026 4218.123 -15.06169 -2540.345
4 -5842.486 49.85133 396.8614 4173.404 -15.03142 -2538.384
5 -5575.582 48.85191 394.5575 4239.510 -15.04722 -2539.218
6 -6029.752 50.72453 390.7748 4161.577 -15.02531 -2539.543
Ensuite, il est essentiel de bien inspecter le modèle avant de se concentrer sur les résultats.
Avec un modèle bayésiens, les choses importantes à vérifier sont :
- Est-ce que notre modèle a bien réussi à reproduire la distribution originale de nos données :
pp_check(m_regression)
Using 10 posterior draws for ppc type 'dens_overlay' by default.
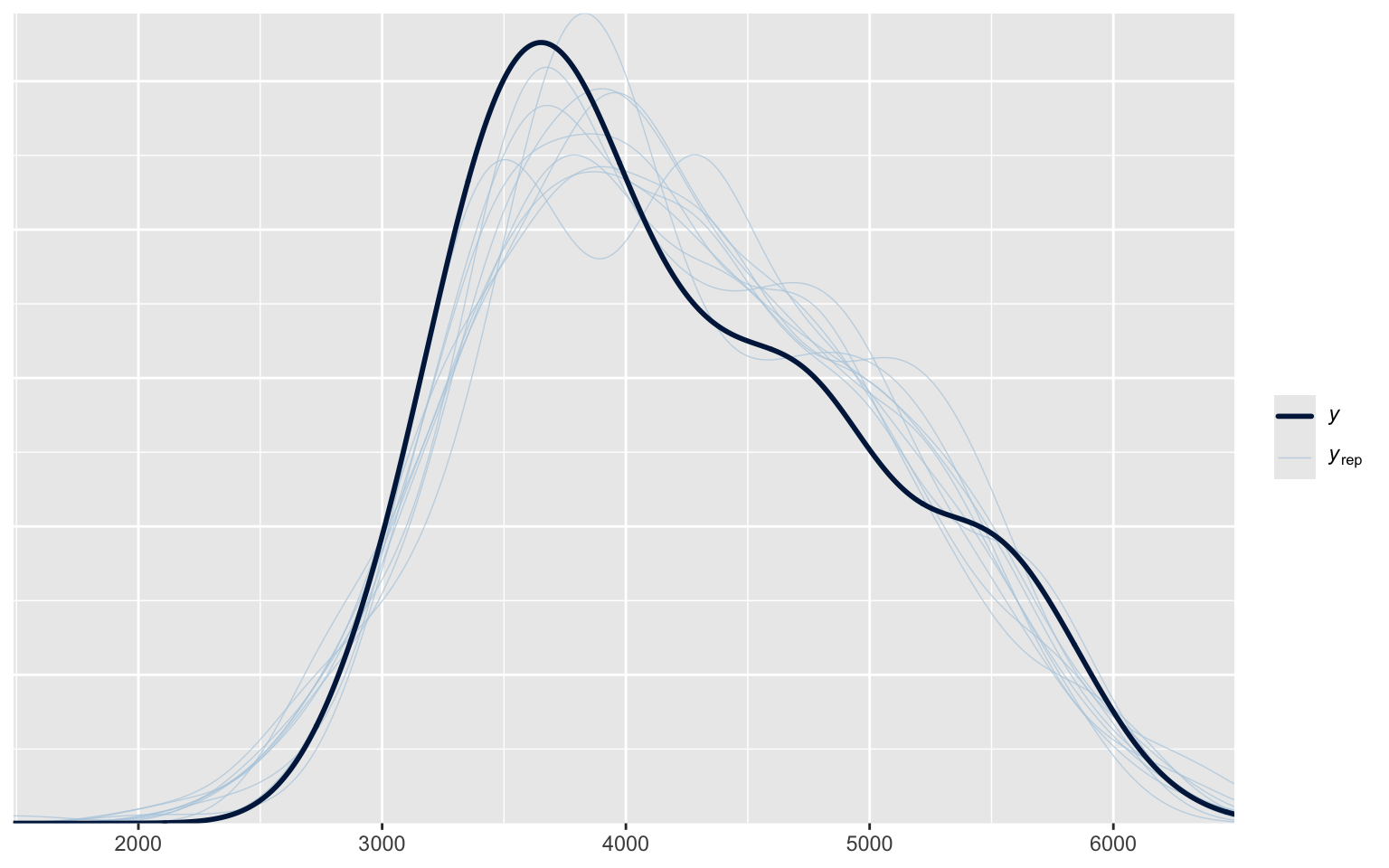
Ce graphique nous illustre la distribution originale du poids de manchots (la ligne noire) et 10 distributions issues de simulations basées sur notre modèle. À première vue, la distribution originale est très bien reproduite
- Ensuite, il convient de s’assurer que le processus d’exploration des valeurs possibles des paramètres ce soit bien déroulé. Pour se faire, la façon classique est de tracer la progression de chacune des chaînes MCMC pour chacun des paramètres :
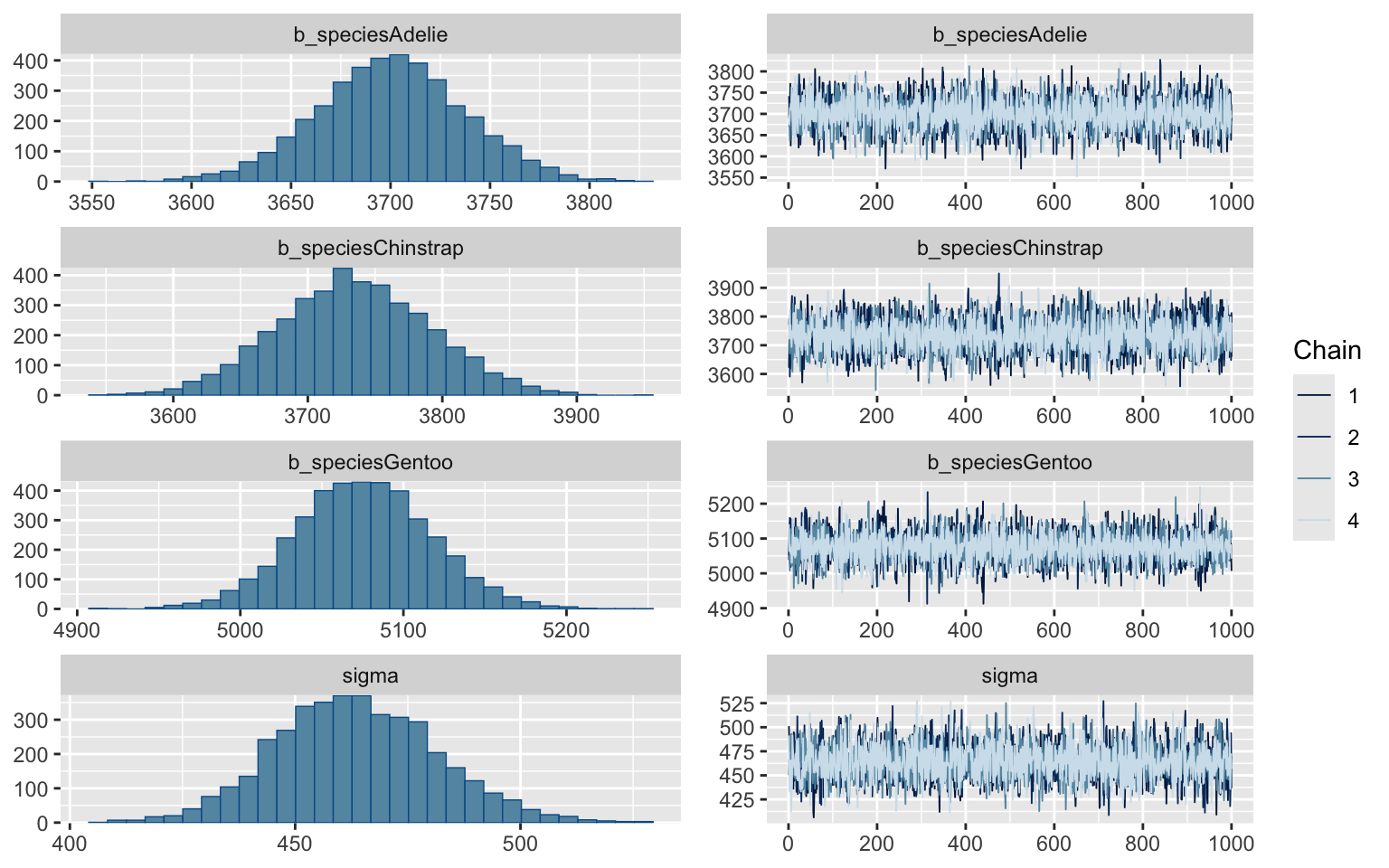
plot(m_regression)
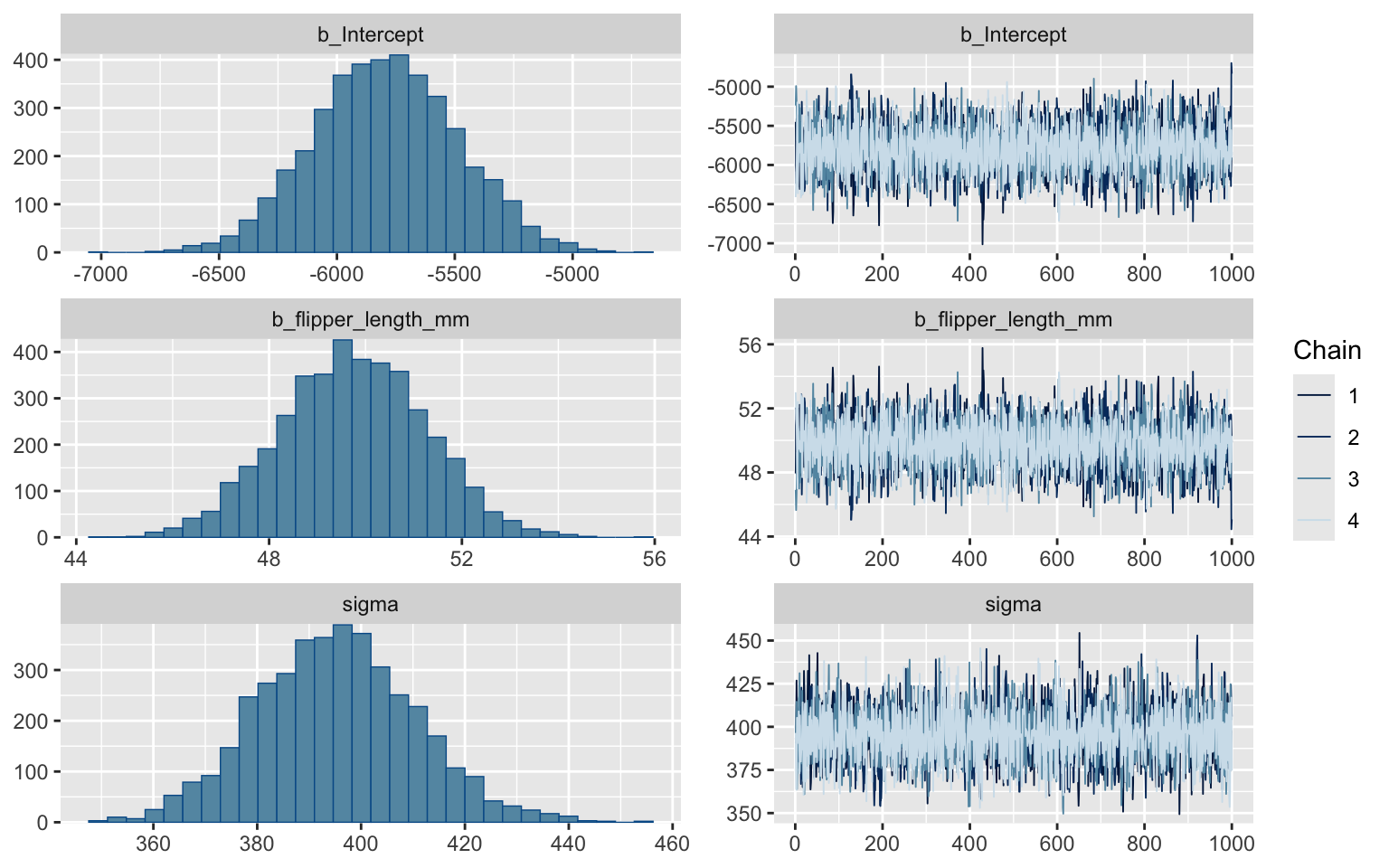
Ici, ce que l’on veut voir dans les panneaux de droite est ce que l’on appelle une “chenille poilue”, bien homogène tout au long des itérations.
Le panneau de gauche nous illustre la distribution de chacun des paramètres. C’est ici où il est avantageux d’avoir chacune des valeurs de paramètres testées par la chaîne. Si pour une raison ou une autre l’incertitude autour d’un paramètre avait suivi autre chose qu’une distribution normale, nous l’aurions automatiquement détecté.
Remarquez qu’ici, le paramètre de l’erreur du modèle (sigma) est distribué relativement normalement puisque les valeurs sont élevées, mais sa distribution sera fréquemment asymétrique, puisqu’un écart-type ne peut pas être négatif. Il ne faut pas vous inquiéter si jamais cela se produit.
- Un autre problème qui pourrait survenir avec notre modèle est que ce dernier pourrait être instable, influencé par une seule ou quelques observations. Nous avons vu dans un atelier précédent qu’il existait une façon d’effectuer la validation croisée dans un modèle bayésien sans ré-ajuster des centaines de modèles, avec l’algorithme PSIS-LOO-CV. Vous vous rappellez peut-être que cet algorithme avait aussi la propriété de détecter du même coup les observations qui influençaient fortement nos résultats, car elles possédaient de fortes valeurs de k de Pareto (i.e. > .7).
loo(m_regression)
Computed from 4000 by 342 log-likelihood matrix.
Estimate SE
elpd_loo -2531.4 13.2
p_loo 2.9 0.3
looic 5062.9 26.5
------
MCSE of elpd_loo is 0.0.
MCSE and ESS estimates assume MCMC draws (r_eff in [0.9, 1.1]).
All Pareto k estimates are good (k < 0.7).
See help('pareto-k-diagnostic') for details.
Donc, ici, pas de problèmes de valeurs influentes.
On peut enfin regarder les sorties de notre modèle :
summary(m_regression)
Family: gaussian
Links: mu = identity; sigma = identity
Formula: body_mass_g ~ flipper_length_mm
Data: pour_regression (Number of observations: 342)
Draws: 4 chains, each with iter = 2000; warmup = 1000; thin = 1;
total post-warmup draws = 4000
Regression Coefficients:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
Intercept -5792.95 304.46 -6381.12 -5189.01 1.00 4014 3133
flipper_length_mm 49.75 1.51 46.75 52.64 1.00 4044 3107
Further Distributional Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma 395.24 15.30 365.84 426.36 1.00 4122 2962
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).
Cette sortie contient beaucoup d’information, mais celles qui nous importent sont que l’ordonnée à l’origine de notre pente est estimée à -5784, et que 95% des chances que la vraie ordonnée à l’origine de la population soit entre -6396 et -5182.
Note
Remarquez que comme l’échantillonnage MCMC est aléatoire, les chiffres présents dans les sorties pourraient être légèrement différents de ceux que je vous rapporte dans le texte, puisqu’ils varieront d’une fois à l’autre.
Je tenterai de contourner le problème en arrondissant les valeurs obtenues, mais les chiffres pourraient tout de même différer légèrement.
De même, la pente estimée dans la relation est de 50 g/mm, avec 95% des chances que la vraie valeur de cette pente soit entre 47 et 53 g/mm.
Enfin, l’écart-type des erreurs (i.e. le bruit résiduel non expliqué par notre modèle) est évalué à 395 g.
Cette sortie nous fournit aussi des preuves supplémentaires que la distribution des paramètres a bien été explorée, puisque les valeur de Rhat sont toutes arrondies à 1.00 (i.e. le modèle a bien convergé) et le nombre d’échantillons indépendants pour chacun des paramètres (Bulk_ESS et Tail_ESS) est très élevé.
Si jamais on veut visualiser cette pente et son incertitude, on peut tracer
son effet marginal en extrayant de notre modèle les prédictions pour chacune
des valeurs de paramètres testée, puis en utilisant la fonction
stat_linerribbon pour extraire l’intervalle de crédibilité (ici à 95%) de l’ensemble
des pentes étudiées. On ajoute, pour bien compléter le portrait, les données
originales au graphique. Ces 5 lignes font donc l’équivalent, dans un
cadre bayésien de ce qu’aurait fait la fonction visreg dans un cadre
fréquentiste.
pour_regression |>
add_epred_draws(m_regression) |>
ggplot(aes(flipper_length_mm, body_mass_g)) +
stat_lineribbon(aes(y = .epred),fill = "gray80",color = "royalblue") +
geom_point(data = pour_regression)
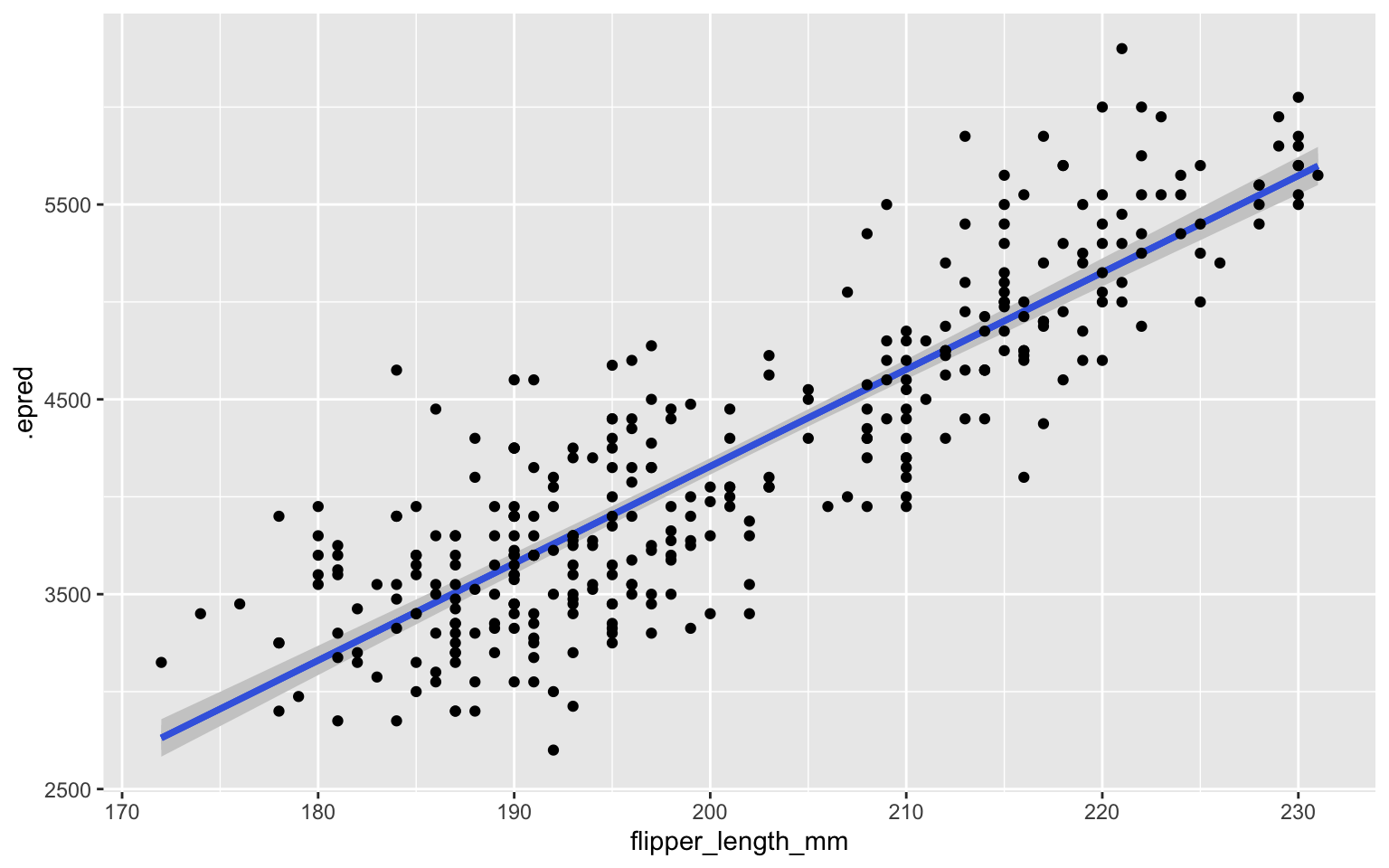
Remarquez qu’au geom_point, il faut fournir notre tableau de données original,
car celui dans la chaîne contient maintenant 4000x chacun des points.
L’approche bayésienne ne cadre pas avec la pratique habituelle des tests d’hypothèses nulles fréquentistes et en ce sens, elle ne fournit pas de valeur de p, elle n’utilise pas de seuils de signification.
Cependant, le fait d’avoir l’ensemble des valeurs testées pour les paramètres permet de
se demander, par exemple, quelle est la probabilité que la vraie pente de la population
soit > 0. Autrement dit, quelle est probabilité que les manchots avec des ailes plus
longues soient plus lourds. brms possède une fonction hypothesis, qui permet
de répondre directement à ces questions, comme ceci :
hypothesis(m_regression, "flipper_length_mm > 0")
Hypothesis Tests for class b:
Hypothesis Estimate Est.Error CI.Lower CI.Upper Evid.Ratio
1 (flipper_length_mm) > 0 49.75 1.51 47.24 52.17 Inf
Post.Prob Star
1 1 *
---
'CI': 90%-CI for one-sided and 95%-CI for two-sided hypotheses.
'*': For one-sided hypotheses, the posterior probability exceeds 95%;
for two-sided hypotheses, the value tested against lies outside the 95%-CI.
Posterior probabilities of point hypotheses assume equal prior probabilities.
Ici, le test un peu futile : il y a 100% des chances que le poids des manchots augmente avec la longueur des ailes.
Warning
Ce 100 % est tout de même conditionnel aux données récoltées et au modèle utilisé.
On peut cependant préciser notre question, et se demander, est-ce que 1 mm d’ailes de plus fournit au moins 50 g de plus à nos manchots ou non.
hypothesis(m_regression, "flipper_length_mm > 50")
Hypothesis Tests for class b:
Hypothesis Estimate Est.Error CI.Lower CI.Upper Evid.Ratio
1 (flipper_length_m... > 0 -0.25 1.51 -2.76 2.17 0.78
Post.Prob Star
1 0.44
---
'CI': 90%-CI for one-sided and 95%-CI for two-sided hypotheses.
'*': For one-sided hypotheses, the posterior probability exceeds 95%;
for two-sided hypotheses, the value tested against lies outside the 95%-CI.
Posterior probabilities of point hypotheses assume equal prior probabilities.
Dans ce cas, le résultat est plus nuancé. Il y a uniquement 42% des chances que cette affirmation soit vraie.
Une chose que les sorties du modèle ne fournissaient pas était une valeur de R2.
Le calcul n’est pas aussi simple, parce qu’entre autres, nous avons 4000 valeurs pour chacun des paramètres. La librairie brms fournit une fonction, nommée bayes_R2 qui permet de tenir compte du fait que nous avons toutes ces valeurs de paramètres. Le calcul sous-jacent est aussi légèrement différent du R2 classique puisqu’il s’assure que toutes les valeurs sont contraintes entre 0 et 1, mais l’important est qu’il s’interprète exactement de la même manière qu’un R2, sauf qu’il fournit aussi une incertitude associées au R2 :
bayes_R2(m_regression)
Estimate Est.Error Q2.5 Q97.5
R2 0.7585594 0.01120369 0.7338805 0.7775618
Ici, la longueur des ailes explique 76% de la variation dans le poids des manchots, et la vraie valeur dans la population a 95% des chances d’être entre 73% et 78% d’explication.
Test de t à deux échantillons
Donc, maintenant, premier saut conceptuel : comment peut-on coder le test de t à deux échantillons dans un cadre de régression?
Notre question pour ce deuxième scénario sera : est-ce que les manchots Adélie ont un poids différent sur l’île Torgersen que sur l’île Biscoe. On pourrait s’attendre à une différence par exemple si une des îles a de meilleures sources d’alimentation, etc.
Donc, nous allons redémarrer du tableau de données original, puis ne garder que les manchots Adélie de ces deux îles. Nous devrons aussi utiliser la fonction fct_drop pour éliminer toute trace de l’île non-utilisée, sinon elle reste présente dans les données d’encodage.
pour_t <-
penguins |>
filter(species == "Adelie") |>
filter(island %in% c("Torgersen","Biscoe")) |>
mutate(island = fct_drop(island)) |>
select(body_mass_g, island) |>
drop_na()
Ensuite, la façon la plus intuitive d’entrer cela dans un modèle de régression est sans doute d’utiliser ce que l’on appelle des variables indicatrices. Dans ce cas, notre variable d’île sera remplacée par autant de variables qu’il y a d’îles, chacune servant à estimer la moyenne pour ce groupe. Pour que cette stratégie fonctionne, il est impératif que l’on élimine l’ordonnée à l’origine du modèle, sinon celui-ci devient non-identifiable.
C’est d’ailleurs le comportement par défaut de plusieurs fonctions (dont lm et brm) : si votre modèle contient
une variable catégorique et que vous demandez d’enlever l’ordonnée à l’origine, R utilisera automatiquement des variables indicatrices.
Mathématiquement, le modèle que nous voudrons ajuster ressemblera donc à ceci : \(\begin{gathered} y_i \sim \mathcal{N}(\mu,\sigma^2)\\ \mu= \beta_{ile[i]} \end{gathered}\) où beta est un vecteur de 2 paramètres, contenant chacun la moyenne d’une île.
Donc, voici à quoi pourrait ressembler le modèle :
m_t <- brm(
body_mass_g ~ 0 + island,
data = pour_t,
cores = 4
)
L’avantage majeur de cette approche est que nos validations, peu importe le contenu du modèle, sont exactement les mêmes :
pp_check(m_t)
Using 10 posterior draws for ppc type 'dens_overlay' by default.
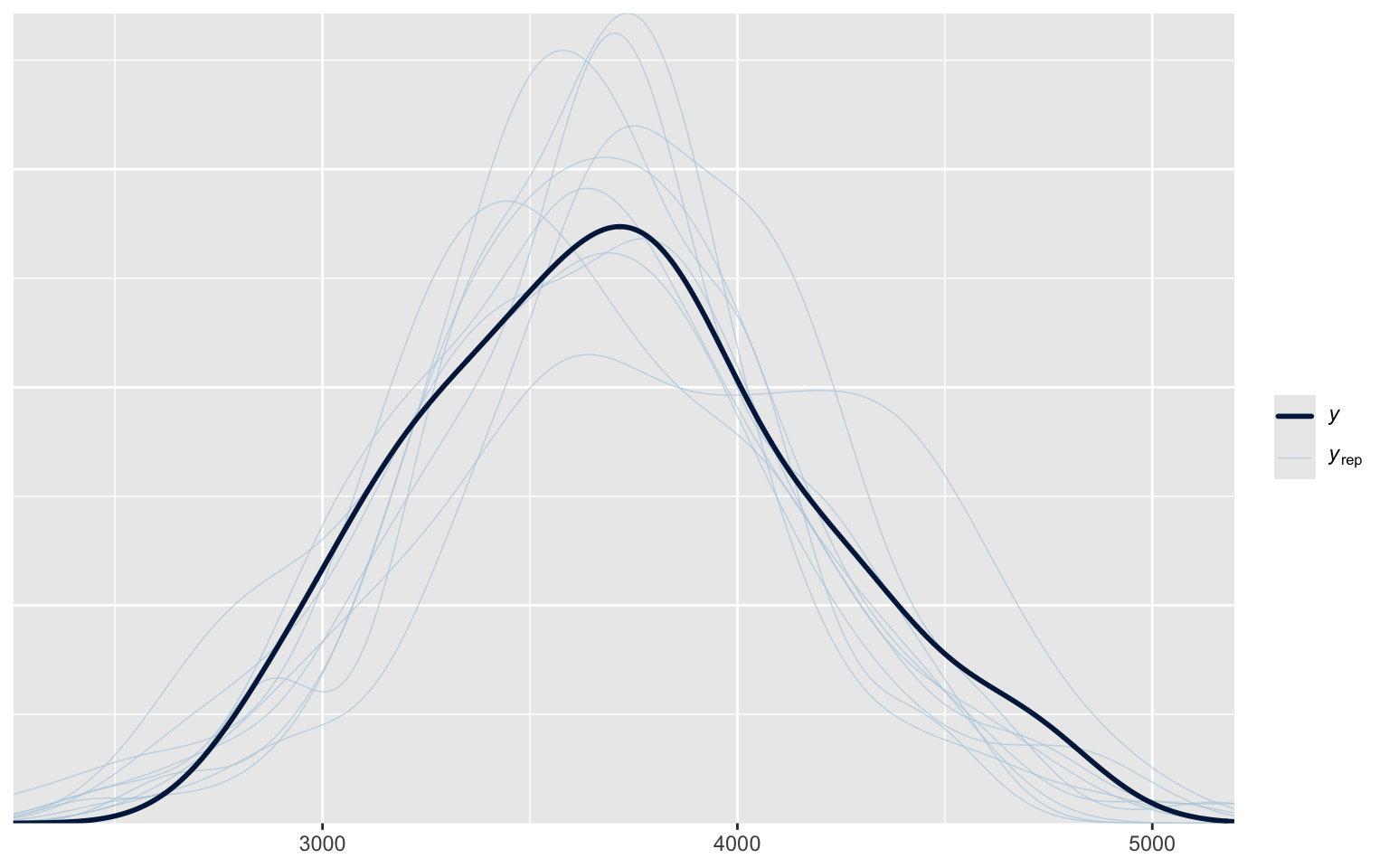
plot(m_t)
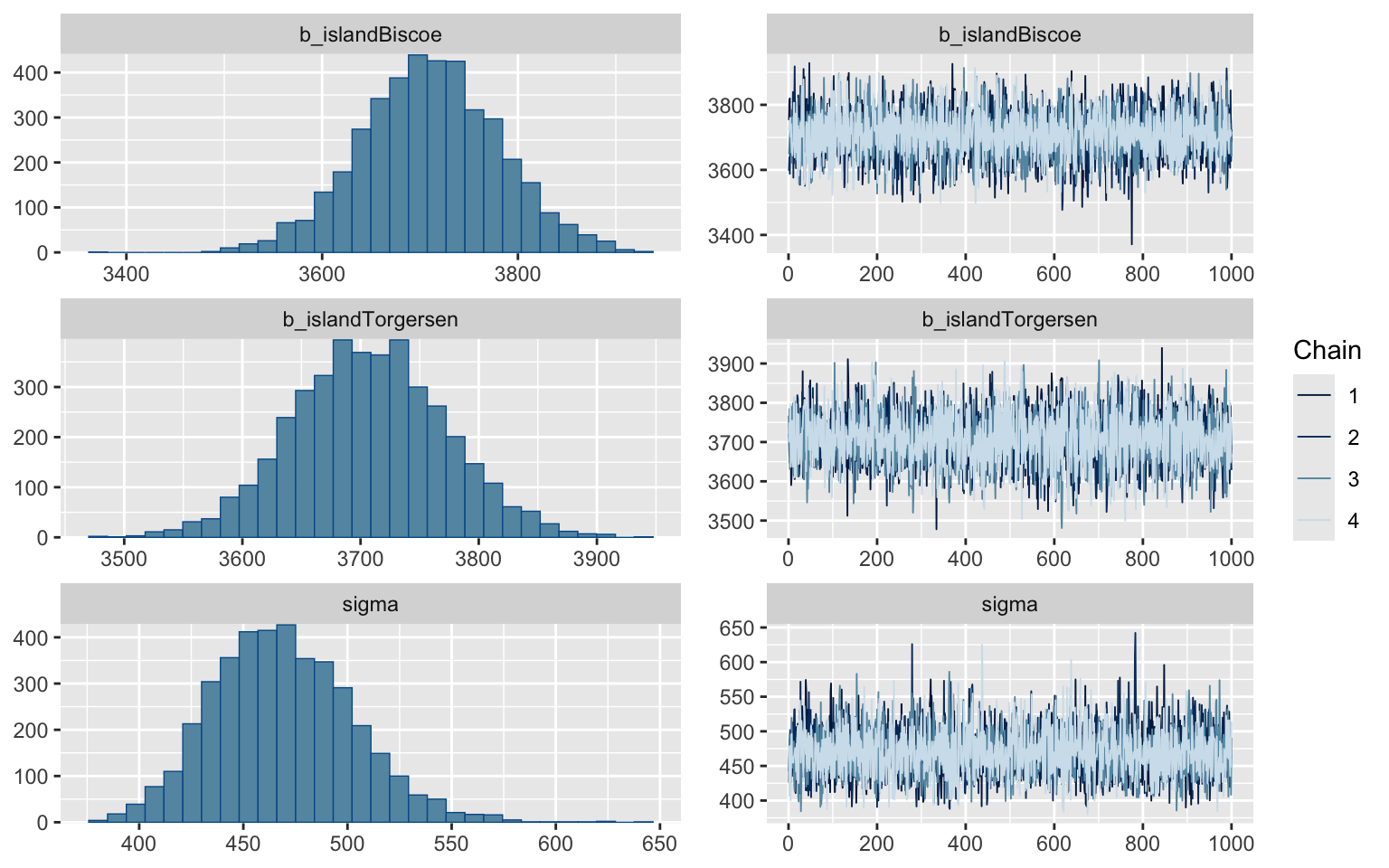
loo(m_t)
Computed from 4000 by 95 log-likelihood matrix.
Estimate SE
elpd_loo -720.2 6.1
p_loo 2.7 0.4
looic 1440.4 12.2
------
MCSE of elpd_loo is 0.0.
MCSE and ESS estimates assume MCMC draws (r_eff in [0.9, 1.1]).
All Pareto k estimates are good (k < 0.7).
See help('pareto-k-diagnostic') for details.
Ici, rien d’inquiétant non plus.
On peut ensuite jeter un coup d’œil à nos résultats :
summary(m_t)
Family: gaussian
Links: mu = identity; sigma = identity
Formula: body_mass_g ~ 0 + island
Data: pour_t (Number of observations: 95)
Draws: 4 chains, each with iter = 2000; warmup = 1000; thin = 1;
total post-warmup draws = 4000
Regression Coefficients:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
islandBiscoe 3709.86 70.85 3568.58 3851.50 1.00 4188 2853
islandTorgersen 3706.84 64.91 3581.53 3837.35 1.00 3962 2813
Further Distributional Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma 469.05 33.79 408.44 541.19 1.00 3924 2615
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).
Ici, on voit que les manchots sur l’île de Biscoe pèsent en moyenne 3708 g, et ceux sur Torgersen en moyenne 3706, avec des intervalles de crédibilité à 95% allant respectivement de 3574 à 3849 g et de 3576 à 3840 g. Autrement dit, il n’y a essentiellement pas de différences de poids entre les manchots des deux îles.
On peut se faire un graphique pour visualiser l’ampleur de cette (minime) différence :
pour_t |>
add_epred_draws(m_t) |>
ggplot(aes(island, body_mass_g)) +
geom_jitter(data = pour_t) +
stat_pointinterval(aes(y = .epred),fill = "gray80",color = "royalblue")
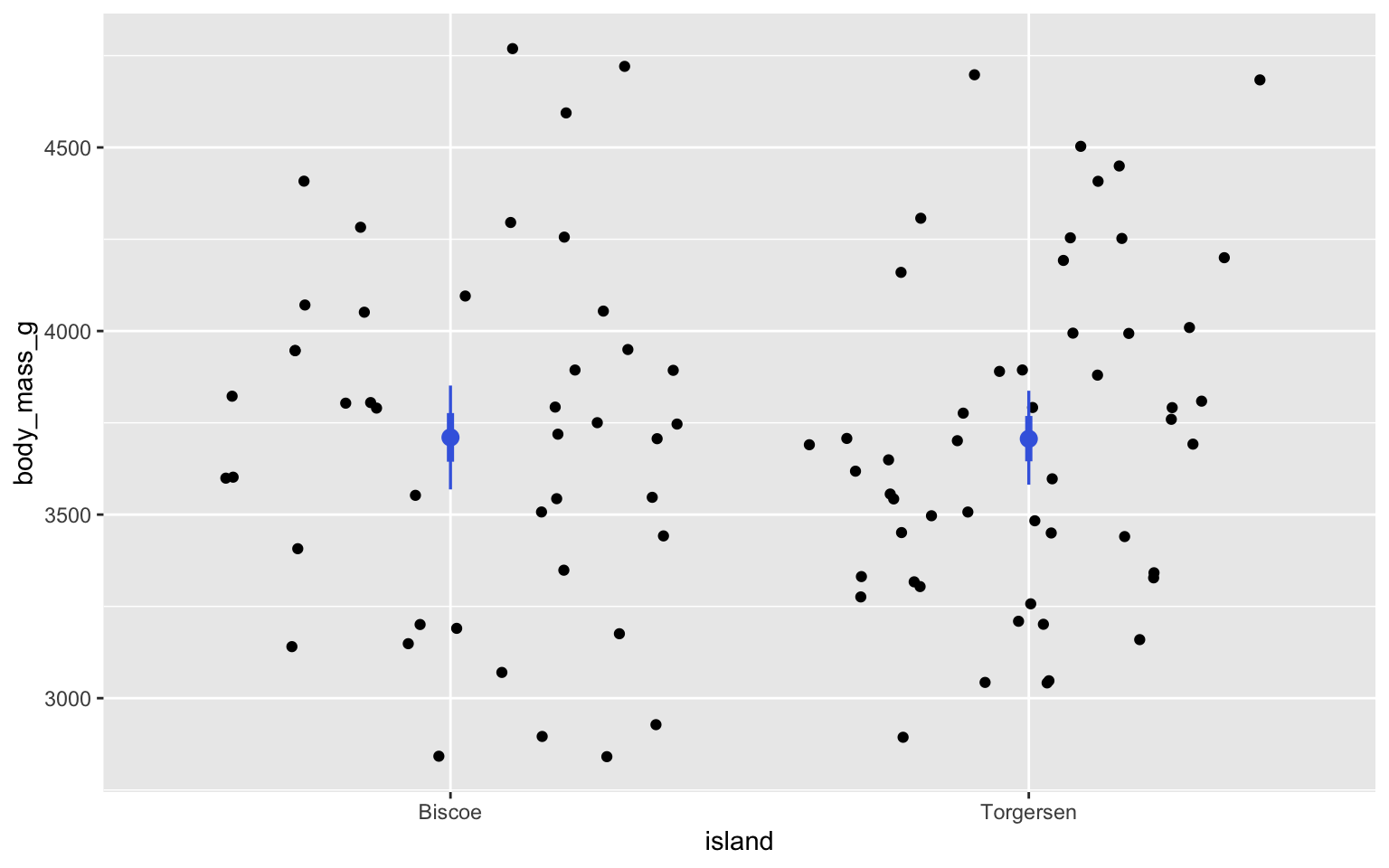
Ici, remarquez que pour une variable catégorique, il faut plutôt utiliser stat_pointinterval pour
illustrer l’intervalle, et que j’ai ajouté du bruit aux points pour que leur densité soit plus claire, qu’ils soient moins empilés.
Encore une fois, puisque nous avons tous les échantillons issus du processus MCMC, on peut demander directement : quelle est la probabilité que les manchots de Biscoe soient plus lourds que ceux de Torgersen?
hypothesis(m_t,"islandBiscoe - islandTorgersen > 0")
Hypothesis Tests for class b:
Hypothesis Estimate Est.Error CI.Lower CI.Upper Evid.Ratio
1 (islandBiscoe-isl... > 0 3.02 95.93 -152.86 157.91 1.07
Post.Prob Star
1 0.52
---
'CI': 90%-CI for one-sided and 95%-CI for two-sided hypotheses.
'*': For one-sided hypotheses, the posterior probability exceeds 95%;
for two-sided hypotheses, the value tested against lies outside the 95%-CI.
Posterior probabilities of point hypotheses assume equal prior probabilities.
Ici, on voit qu’il y avait seulement 51% des combinaisons de paramètres où les manchots de Biscoe étaient estimés comme plus lourds que ceux de Torgersen.
Test de F
Un autre test classique de l’introduction aux biostatistiques est sans doute le test de F. Celui-ci est bien sûr imbriqué dans les calculs de l’ANOVA et a déjà été utilisé à une époque comme pré-test pour déterminer quel test de t utiliser, mais il peut être aussi utilisé de façon indépendante, pour déterminer si un groupe est plus variable qu’un autre groupe.
Dans le scénario précédent où l’on regardait les différences de poids des manchots entre deux îles, nous nous étions arrêtés à la moyenne. Cependant, on pourrait aussi se demander si les poids sont plus variables (i.e. hétérogènes) dans une île qu’une autre, peut-être à cause de l’hétérogénéité du paysage?
On peut explorer cette question avec exactement les mêmes données et le même modèle, à une petite différence près : nous allons spécifier que sigma aussi peut varier selon les îles.
Avec la notation proposée ci-haut, il est très intuitif de faire ce changement :
\[\begin{gathered} y_i \sim \mathcal{N}(\mu,\sigma^2)\\ \mu= \beta_{ile[i]} \\ \sigma= \delta_{ile[i]} \end{gathered}\]Nous avons donc un vecteur de 2 paramètres additionnels, delta, qui contient l’écart-type des erreurs pour chacune des îles
Dans brms, nous devrons donc spécifier deux formules, une pour définir ce qui peut influencer la moyenne du poids des manchots, et une qui va spécifier ce qui peut influencer la variabilité du poids. Remarquez qu’il faut ensuite emballer ces deux formules par la fonction bf, pour Bayes Formula.
m_f <- brm(
bf(
body_mass_g ~ 0 + island,
sigma ~ 0 + island
),
data = pour_t,
cores = 4
)
Encore une fois, nos vérifications ne trouvent rien d’inquiétant :
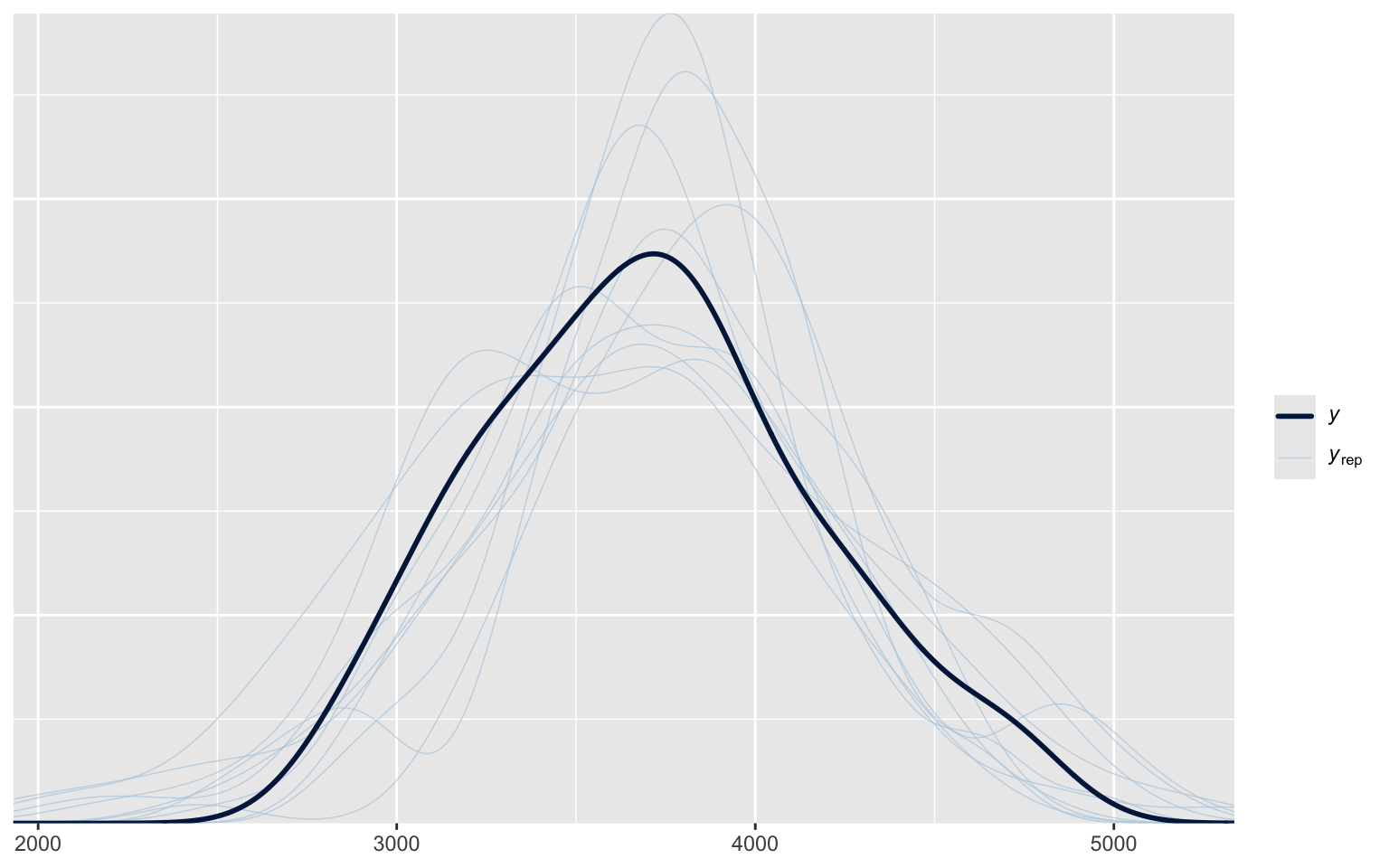
pp_check(m_f)
Using 10 posterior draws for ppc type 'dens_overlay' by default.
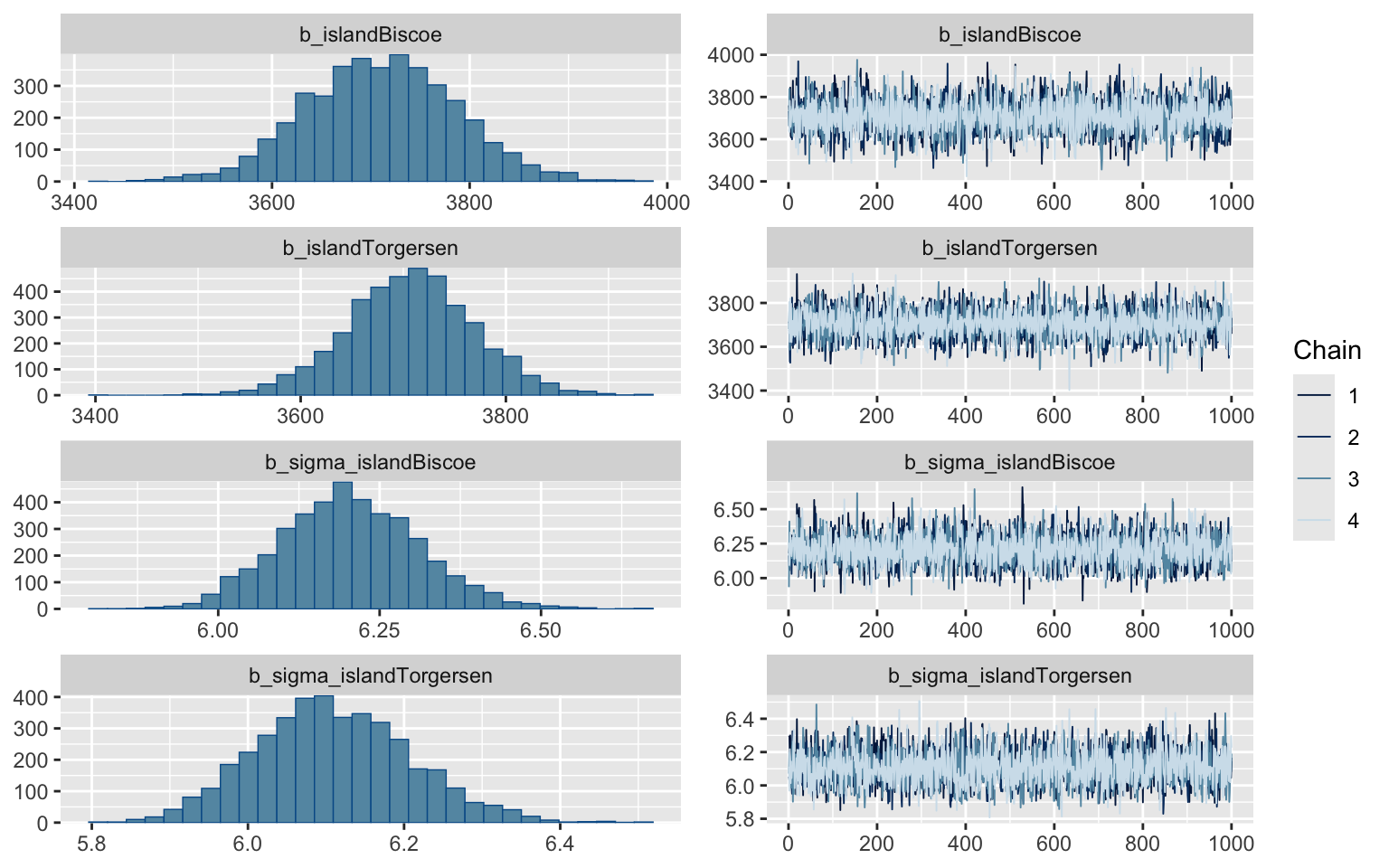
plot(m_f)
loo(m_f)
Computed from 4000 by 95 log-likelihood matrix.
Estimate SE
elpd_loo -721.0 6.1
p_loo 3.4 0.5
looic 1442.0 12.2
------
MCSE of elpd_loo is 0.0.
MCSE and ESS estimates assume MCMC draws (r_eff in [0.9, 1.2]).
All Pareto k estimates are good (k < 0.7).
See help('pareto-k-diagnostic') for details.
summary(m_f)
Family: gaussian
Links: mu = identity; sigma = log
Formula: body_mass_g ~ 0 + island
sigma ~ 0 + island
Data: pour_t (Number of observations: 95)
Draws: 4 chains, each with iter = 2000; warmup = 1000; thin = 1;
total post-warmup draws = 4000
Regression Coefficients:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS
islandBiscoe 3710.36 76.64 3561.91 3860.11 1.00 3953
islandTorgersen 3707.19 62.91 3582.56 3831.37 1.00 4648
sigma_islandBiscoe 6.20 0.11 6.00 6.43 1.00 4240
sigma_islandTorgersen 6.11 0.10 5.93 6.32 1.00 4421
Tail_ESS
islandBiscoe 2725
islandTorgersen 3087
sigma_islandBiscoe 3332
sigma_islandTorgersen 3106
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).
Nos estimations des poids moyens n’ont pas vraiment bougé, étant maintenant à 3712 g pour Biscoe et 3707 g pour Torgersen. Cependant, nous avons maintenant deux paramètres pour sigma, soit un pour chacune des îles (nous avons ici aussi utilisé des variables d’index). On constate que la variabilité sur Biscoe est légèrement plus élevée sur Biscoe que Torgersen (6.20 vs 6.11) mais que comparé à l’incertitude sur ces paramètres (respectivement 6-6.43 vs 5.92-6.32), cette différence n’est pas très grande.
Note
Si jamais vous trouvez que nos estimations pour sigma sont maintenant minuscules comparées aux modèles précédents, vous avez tout à fait raison.
Sans nous avertir,
brma travaillé les paramètres de variabilité à l’échelle logarithmique, puisque ces derniers ne doivent jamais être négatifs.Ce que
brmsa ajusté était en fait :
\[\begin{gathered} y_i \sim \mathcal{N}(\mu,\sigma^2)\\ \mu= \beta_{ile[i]} \\ \sigma= exp(\delta_{ile[i]}) \end{gathered}\]
Si on passe les valeurs estimées (les B de notre équation) à l’exponentielle
exp(6.20)[1] 492.749exp(6.11)[1] 450.3387on retrouve des valeurs très comparables à ce que l’on avait précédemment.
Encore une fois, on peut se poser des questions probabilistes et obtenir des réponses claires, faciles à interpréter. Par exemple, si on se demande quelle est la probabilité que le poids des manchots de Biscoe soit effectivement plus variable que celui sur Torgersen, on peut le calculer comme ceci :
hypothesis(m_f,"sigma_islandBiscoe - sigma_islandTorgersen > 0")
Hypothesis Tests for class b:
Hypothesis Estimate Est.Error CI.Lower CI.Upper Evid.Ratio
1 (sigma_islandBisc... > 0 0.1 0.14 -0.14 0.33 2.99
Post.Prob Star
1 0.75
---
'CI': 90%-CI for one-sided and 95%-CI for two-sided hypotheses.
'*': For one-sided hypotheses, the posterior probability exceeds 95%;
for two-sided hypotheses, the value tested against lies outside the 95%-CI.
Posterior probabilities of point hypotheses assume equal prior probabilities.
Et savoir que malgré tout, il y a 72% des chances que les manchots de Biscoe soient effectivement plus variables.
Tip
Si jamais vous avez plus de 2 groupes à explorer, l’ensemble de la méthode reste exactement identique. Nul besoin d’utiliser une autre procédure (vs. par exemple le test de Levene).
Remarquez que ce modèle pourrait remplacer aussi un appel à gls, si jamais vous
savez que les variances sont différentes entre les groupes (hétéroscédastique)
et que vous voulez en tenir compte dans votre modèle.
ANOVA
Parmi les autres classiques des cours d’introduction aux biostatistiques, nous ne pouvons évidemment pas passer outre l’ANOVA, conçue pour comparer la moyenne de 2 groupes ou plus (mais qui pour mêler les étudiants est une analyse de la variance!)
Ici, l’ANOVA perd toute personnalité ou spécificité qui pouvait lui être associée. Tout d’abord, pour la petite histoire, sachez que la fonction de base de R pour l’ANOVA, soit aov, est décrite par ses auteurs comme essentiellement identique à lm, dont elle présente seulement les résultats de façon différente :
This provides a wrapper to lm for fitting linear models to balanced or unbalanced experimental designs.
The main difference from lm is in the way print, summary and so on handle the fit: this is expressed in the traditional language of the analysis of variance rather than that of linear models.
De notre point de vue de régression, l’approche est ABSOLUMENT identique à notre conversion du test de t. En tout points!
\[\begin{gathered} y_i \sim \mathcal{N}(\mu,\sigma^2)\\ \mu= \beta_{ile[i]} \end{gathered}\]Mais ici, on mettra plus de 2 valeurs dans notre vecteur beta, c’est tout!
Pour la forme, nous prendrons tout de même le temps de se poser une nouvelle version, soit : est-ce que le poids de nos 3 espèces de manchots diffère. Est-ce que le poids est une caractéristique qui les distingue?
On se créera donc un petit tableau pour travailler, qui contiendra uniquement le poids et l’espèce pour chacun des individus :
pour_anova <-
penguins |>
select(body_mass_g, species) |>
drop_na()
Puis, on passe le tout à la fonction brm :
m_anova <- brm(
body_mass_g ~ 0 + species,
data = pour_anova,
cores = 4
)
On valide, en ne trouvant encore rien d’inquiétant :
pp_check(m_anova)
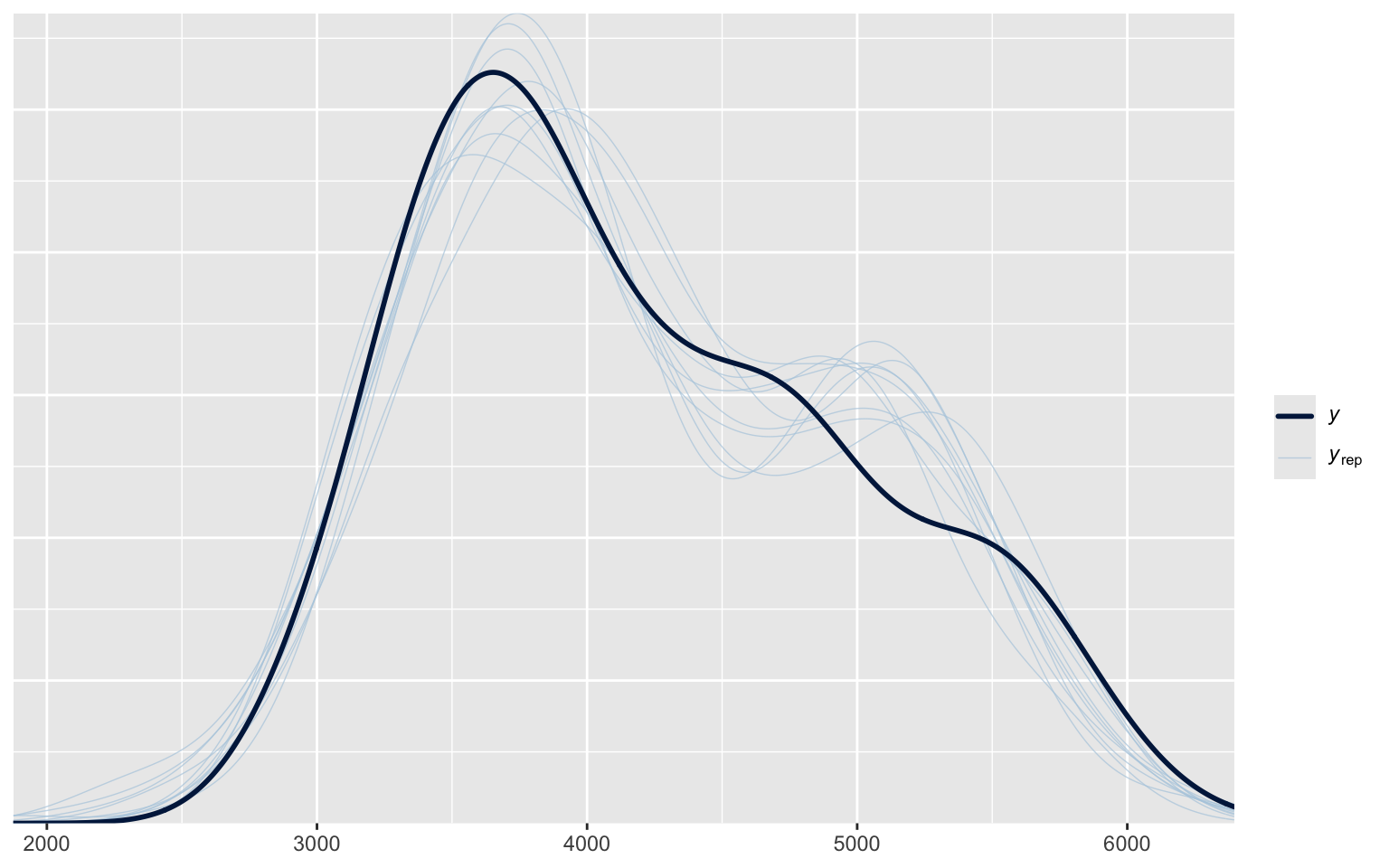
Using 10 posterior draws for ppc type 'dens_overlay' by default.
plot(m_anova)
loo(m_anova)
Computed from 4000 by 342 log-likelihood matrix.
Estimate SE
elpd_loo -2586.2 11.4
p_loo 3.6 0.3
looic 5172.4 22.8
------
MCSE of elpd_loo is 0.0.
MCSE and ESS estimates assume MCMC draws (r_eff in [1.0, 1.3]).
All Pareto k estimates are good (k < 0.7).
See help('pareto-k-diagnostic') for details.
Et enfin, on peut explorer nos résultats :
summary(m_anova)
Family: gaussian
Links: mu = identity; sigma = identity
Formula: body_mass_g ~ 0 + species
Data: pour_anova (Number of observations: 342)
Draws: 4 chains, each with iter = 2000; warmup = 1000; thin = 1;
total post-warmup draws = 4000
Regression Coefficients:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
speciesAdelie 3700.24 37.26 3626.47 3773.06 1.00 4534 3102
speciesChinstrap 3734.56 56.97 3624.66 3846.59 1.00 4654 2827
speciesGentoo 5075.62 41.72 4994.77 5158.87 1.00 5164 2848
Further Distributional Parameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sigma 463.50 17.97 429.89 499.84 1.00 4633 2915
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).
On peut voir clairement que les Gentoo sont plus massifs que les autres espèces, par presque 1300 g, et que cette différence est beaucoup plus importante que l’incertitude associée aux paramètres.
Si on a un doute sur la différence entre les Adelie et les Chinstrap, on peut se demander : quelle est la probabilité que les Chinstrap soient en moyenne effectivement plus lourds que les Adelie :
hypothesis(m_anova,"speciesChinstrap > speciesAdelie")
Hypothesis Tests for class b:
Hypothesis Estimate Est.Error CI.Lower CI.Upper Evid.Ratio
1 (speciesChinstrap... > 0 34.32 67.53 -77.9 142.57 2.29
Post.Prob Star
1 0.7
---
'CI': 90%-CI for one-sided and 95%-CI for two-sided hypotheses.
'*': For one-sided hypotheses, the posterior probability exceeds 95%;
for two-sided hypotheses, the value tested against lies outside the 95%-CI.
Posterior probabilities of point hypotheses assume equal prior probabilities.
Comme l’approche par la régression insiste surtout sur l’estimation de paramètres, elle ne permet pas de répondre directement à la question : Est-ce qu’ajouter la variable d’espèce améliore notre capacité à prédire le poids des manchots?
Si on veut vraiment répondre à cette question, on peut se construire un petit modèle alternatif, qui ne contient pas la variable d’espèce (donc ici, uniquement l’ordonnée à l’origine) :
m_sans_ile <- brm(
body_mass_g ~ 1,
data = pour_anova,
cores = 4
)
Et ensuite comparer la qualité des prédictions de ces deux modèles en se basant sur la validation croisée :
loo_compare(
loo(m_anova),
loo(m_sans_ile)
)
elpd_diff se_diff
m_anova 0.0 0.0
m_sans_ile -187.4 12.4
Ici, le modèle sans l’espèce est clairement moins bon que le modèle avec. Si on se fie au terme d’erreur, le modèle sans l’île est à plus de 15 erreurs-type de celui avec l’île. En sachant que les différences de plus de 4 erreurs-types sont habituellement considérées comme importantes, cette différence est très claire!
Le test de chi-carré
Le dernier test classique dont nous discuterons aujourd’hui est le test de chi-carré (ou chi-deux pour certains).
Si vous vous rappelez, ce test sert à savoir si les proportions des niveaux d’une variable catégorique sont différentes selon les niveaux d’une seconde variable catégorique.
Du point de vue de l’enseignant, une des choses les plus frustrantes à propos de ce test est qu’après avoir insisté pendant des semaines sur l’importance de rapporter et d’interpréter les tailles d’effet, l’outil classique pour comparer les proportions n’en propose aucun. On obtient une valeur de p, et c’est tout.
Heureusement, comme tous les autres tests classiques, ce dernier peut facilement être recodé dans un cadre de régression.
La question qui nous intéressera ici sera de savoir si le ratio mâle-femelle des manchots capturés varie entre les îles. Cette question pourrait être d’intérêt par exemple si on sait en amont que le stress affecte ce ratio et que l’on soupçonne certaines îles d’avoir été exposées à davantage de facteurs de stress que d’autres.
Nous aurons donc besoin de conserver uniquement 2 variables catégoriques :
pour_chi_carre <-
penguins |>
select(island, sex) |>
drop_na()
Et ensuite, nous voudrons expliquer le sexe par l’île, en utilisant à nouveau des variables indicatrices, puisqu’elles sont plus faciles à interpréter. Par contre, la variable à expliquer, le sexe, n’est plus du tout décrite par la distribution normale. Elle est plutôt décrite par la distribution catégorique, une extension de la distribution binomiale lorsque nous avons plus de deux groupes.
Donc :
\(y_i \sim \mathcal{Categorical}(\pi_{1,i},\pi_{2,i},\dots,\pi_{K,i})\\\) Ici, nous n’aurons que deux valeurs de pi, une pour chaque sexe, mais le modèle fonctionnerait pour autant de catégoriques que nécessaire.
Remarquez qu’il est clair dans cette notation pourquoi nous n’avons plus de terme “d’erreur” dans notre modèle (i.e. sigma). Contrairement à la distribution normale, la distribution catégorique (tout comme la Bernoulli ou la Binomiale) ne possède pas de paramètre de dispersion indépendant : la variabilité des données est entièrement déterminée par les probabilités comme tel.
Par contre, comme notre prédicteur linéaire doit pouvoir aller de -Inf à +Inf et que les probabilités ne peuvent clairement pas le faire, nous devrons aussi choisir une fonction de lien pour passer de un à l’autre, ici, la fonction softmax : \(\pi_{k,i} = \frac{exp(\eta_{k,i})}{\sum_{j=1}^{K}exp(\eta_{j,i})}\) Remarquez que cette fonction de lien normalise aussi pour que la somme des probabilités soit toujours égale à 1.
Et enfin, on peut spécifier la partie déterministe du modèle, soit : \(\eta_{k,i} = \beta_{k,ile[i]}\)
Autrement dit, nous un beta par île, comparant le ratio mâle-femelle.
Si nous avions eu plus de 2 niveaux à notre variable expliquée, nous aurions eu, pour chaque île, k-1 paramètres, soit toutes les comparaisons à notre catégorie de référence.
Note
Bien qu’il nous soit possible d’utiliser les variables indicatrices pour l’île, la variable expliquée sera cependant convertie en dummy coding, où une valeur sera choisie comme référence et les autres comparées à celle-ci. De par la nature de modélisation de ratios de cette technique, il n’est pas vraiment possible de faire autrement.
Bien que la notation ci-haut cadre mieux avec le reste de l’atelier, l’étape intermédiaire peut être tout de même un peu difficle à se mettre en tête. Sachez que pour plus que claré, cette formulation est entièrement équivalent, et clarifie le fait que chacun des niveaux de la variable expliquée est comparé à un niveau de référence :
\[\begin{gathered} y_i \sim \text{Categorical}(\pi_{1,i}, \pi_{2,i}, \dots, \pi_{K,i}) \\ \ln \left( \frac{\pi_{k,i}}{\pi_{\text{reference},i}} \right) = \beta_{k,ile[i]} \end{gathered}\]m_chi_carre <-
brm(
sex ~ 0 + island,
family = categorical(),
data = pour_chi_carre
)
Après avoir ajusté le modèle, il faut comme toujours l’inspecter. Mais remarquez qu’ici, même si on ajuste techniquement un GLM, les vérifications de base demeurent les mêmes.
pp_check(m_chi_carre)
Using 10 posterior draws for ppc type 'dens_overlay' by default.
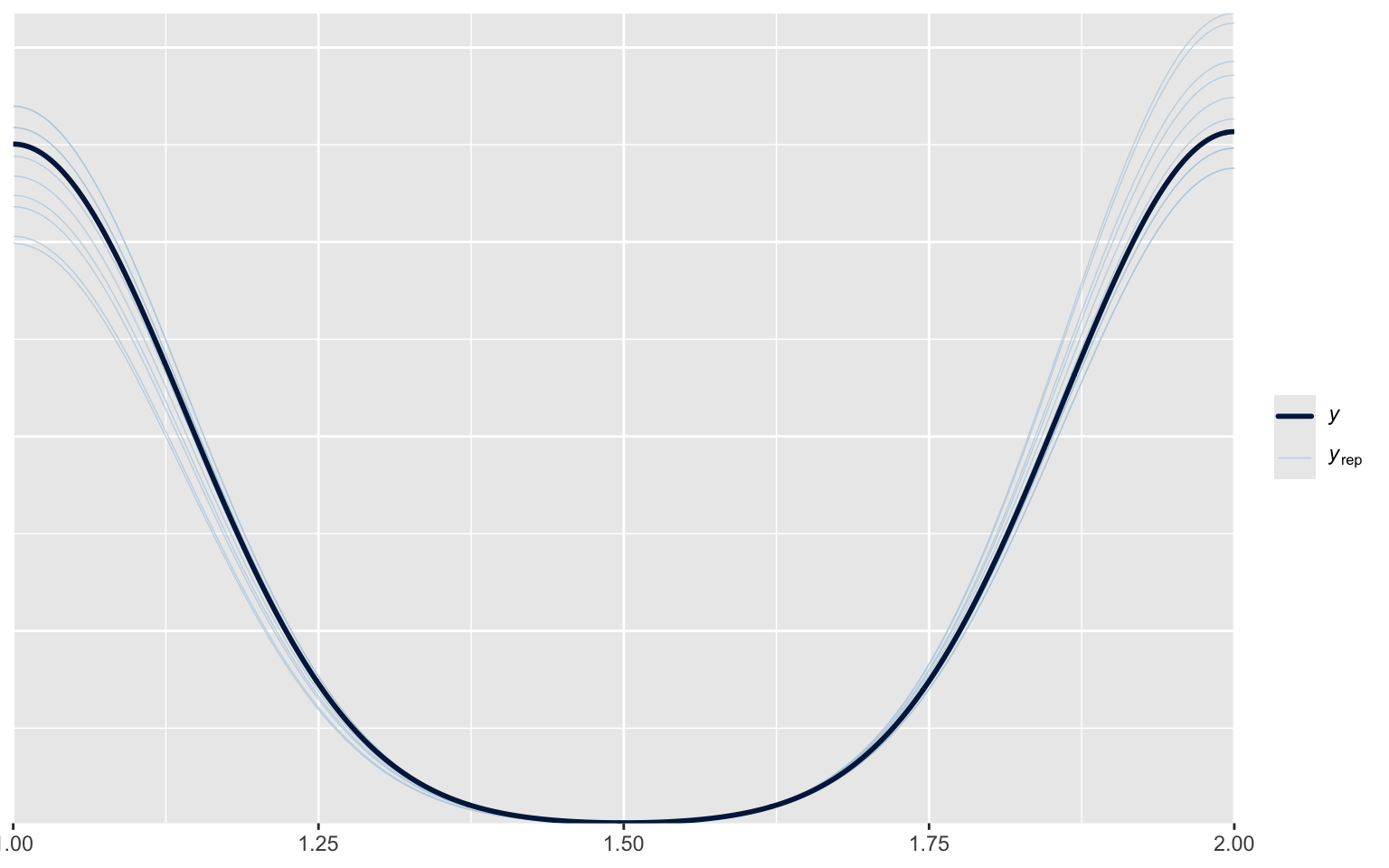
Ici, comme notre modèle ne prédit que des catégories, le lissage n’est pas nécessairement la meilleure façon d’explorer la distribution.
pp_check(m_chi_carre, type = "bars_grouped",group="island")
Using 10 posterior draws for ppc type 'bars_grouped' by default.
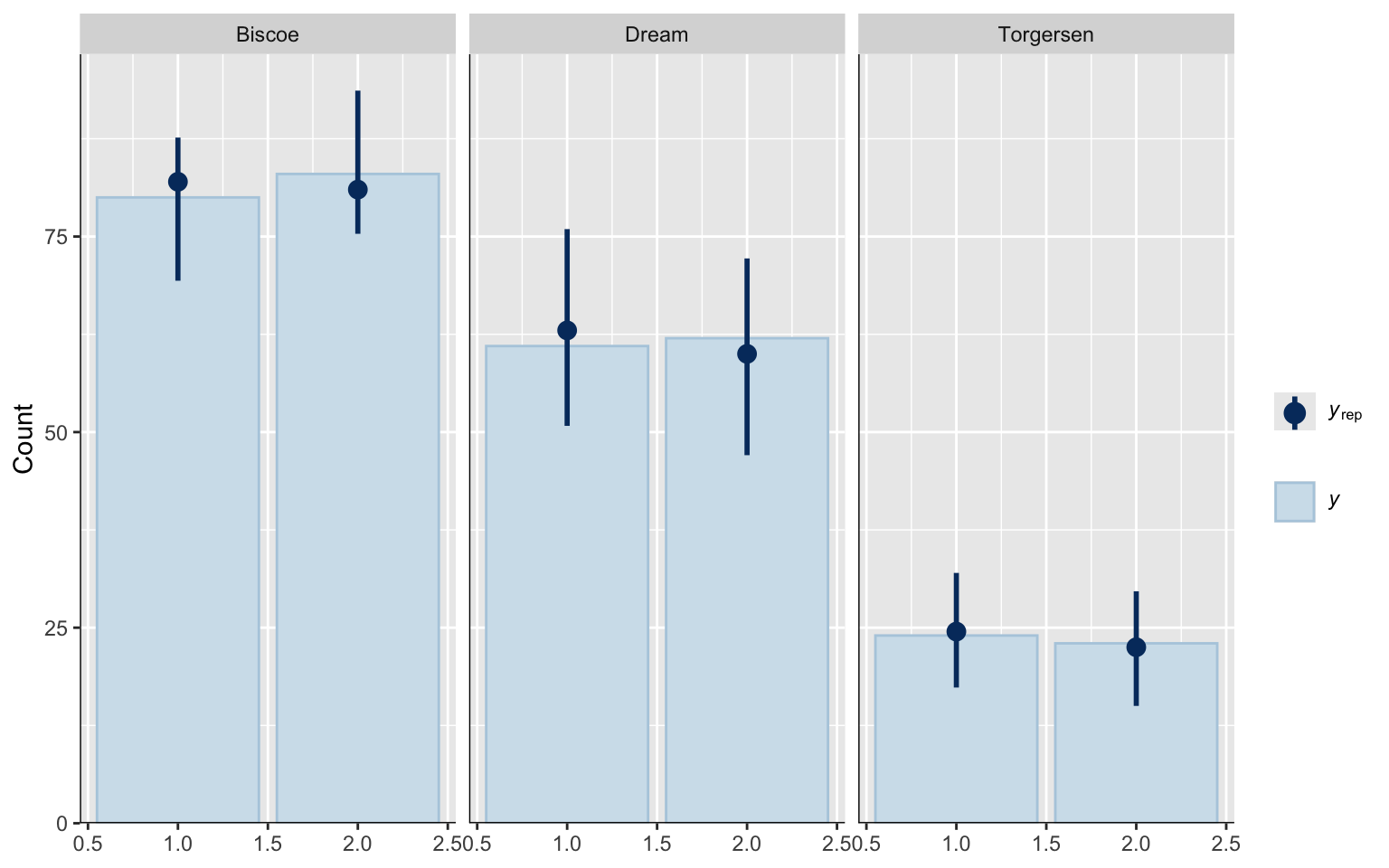
Par contre, en les séparant en bandes par groupes, c’est beaucoup plus clair que l’on reproduit adéquatement la distribution originale.
plot(m_chi_carre)
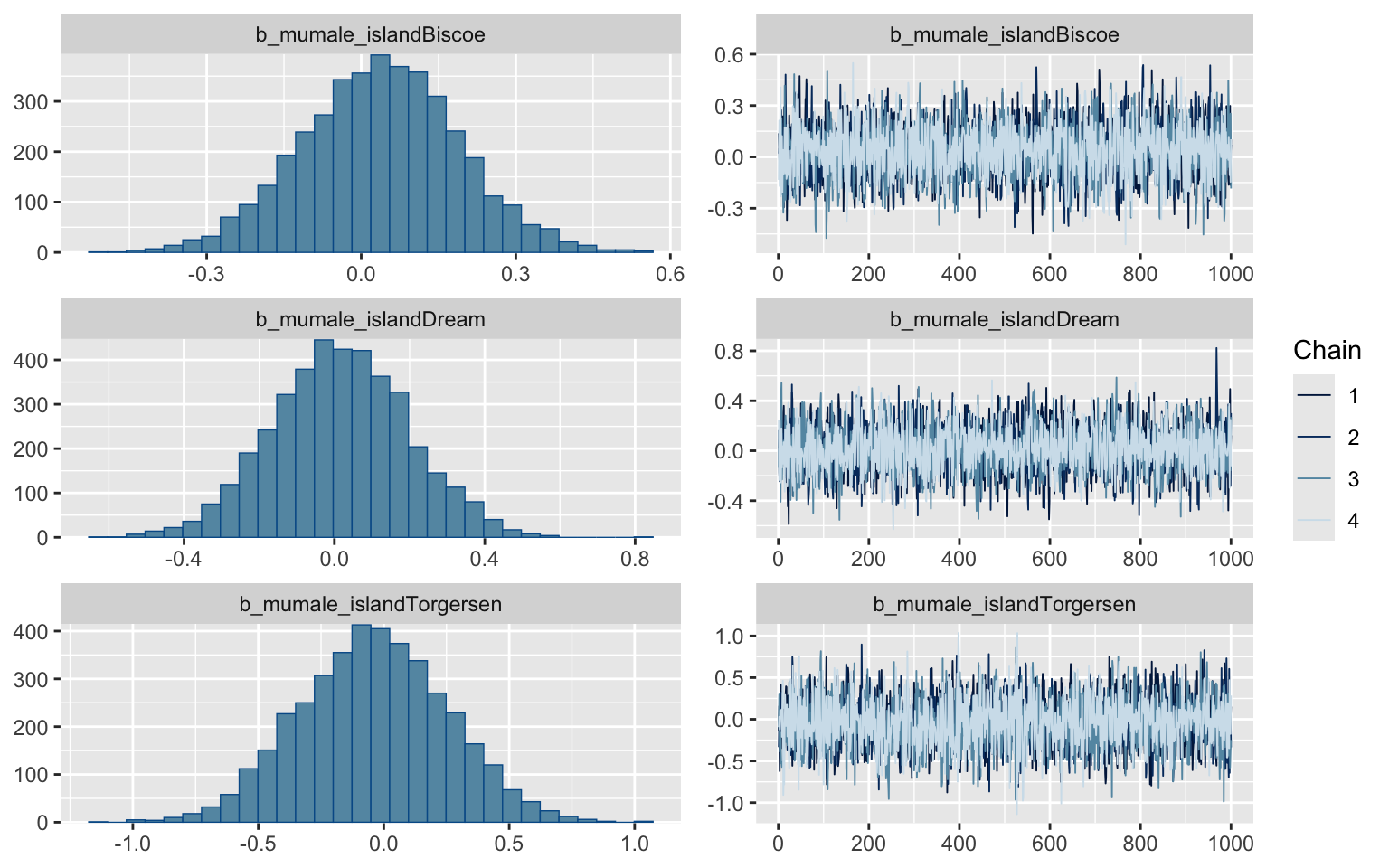
loo(m_chi_carre)
Computed from 4000 by 333 log-likelihood matrix.
Estimate SE
elpd_loo -233.8 0.3
p_loo 3.0 0.1
looic 467.6 0.6
------
MCSE of elpd_loo is 0.0.
MCSE and ESS estimates assume MCMC draws (r_eff in [0.9, 1.0]).
All Pareto k estimates are good (k < 0.7).
See help('pareto-k-diagnostic') for details.
summary(m_chi_carre)
Family: categorical
Links: mumale = logit
Formula: sex ~ 0 + island
Data: pour_chi_carre (Number of observations: 333)
Draws: 4 chains, each with iter = 2000; warmup = 1000; thin = 1;
total post-warmup draws = 4000
Regression Coefficients:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS
mumale_islandBiscoe 0.04 0.15 -0.27 0.35 1.00 3691
mumale_islandDream 0.02 0.18 -0.33 0.37 1.00 3706
mumale_islandTorgersen -0.04 0.30 -0.61 0.54 1.00 3870
Tail_ESS
mumale_islandBiscoe 2805
mumale_islandDream 2957
mumale_islandTorgersen 2992
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).
Ici, les estimations de paramètres demandent cependant un peu plus de réflexion.
Les estimations de paramètres fournies par brms sont celles des beta dans
nos équations ci-haut. On peut récupérer le ratio des cotes en prenant
l’exponentielle des estimations de paramètres fournies.
Pour le premier paramètre, mumale_islandBiscoe :
exp(0.04)
[1] 1.040811
Sur l’île de Biscoe, il y a en moyenne 1,04 mâle pour chaque femelle.
exp(-0.04)
[1] 0.9607894
Sur Torgersen, c’est 0,96 mâle pour une femelle
Mais notre incertitude est très grande sur ces ratios.
Si on voulait savoir la probabilité que le ratio mâle-femelle soit plus élevé sur l’île de Biscoe que sur l’île de Torgersen, on pourrait poser la question comme ceci :
hypothesis(m_chi_carre,"mumale_islandBiscoe>mumale_islandTorgersen")
Hypothesis Tests for class b:
Hypothesis Estimate Est.Error CI.Lower CI.Upper Evid.Ratio
1 (mumale_islandBis... > 0 0.08 0.33 -0.46 0.62 1.45
Post.Prob Star
1 0.59
---
'CI': 90%-CI for one-sided and 95%-CI for two-sided hypotheses.
'*': For one-sided hypotheses, the posterior probability exceeds 95%;
for two-sided hypotheses, the value tested against lies outside the 95%-CI.
Posterior probabilities of point hypotheses assume equal prior probabilities.
Il y a donc uniquement 60% des chances que le ratio mâle/femelle sur l’île de Biscoe soit effectivement plus élevé que sur l’île de Torgersen.
Vers l’infini et plus loin encore
Le plus épatant est que la même fonction peut aussi nous permettre d’ajuster un GLMM avec des termes de lissage, sans même sourciller.
Par exemple, si on voulait savoir si on peut deviner le sexe (régression logistique) d’un individu à partir d’une courbe non-paramétrique basée sur l’épaisseur du bec, avec un effet aléatoire sur l’identité de l’île (voir ci-haut), on pourra le faire tout aussi facilement :
pour_gros_modele <-
penguins |>
select(sex, island, bill_depth_mm) |>
drop_na()
m_gros <-
brm(
sex ~ s(bill_depth_mm) + (1|island),
data = pour_gros_modele,
family = bernoulli(),
cores = 4,
)
Il se peut qu’à ce point ci, vous obteniez des avertissements comme quoi notre
modèle a produit des transitions divergentes. Comme celui-ci est techniquement plus
complexe, son espace de paramètres est un peu plus complexe à explorer et il
faudrait probablement ajuster adapt_delta pour lui permettre de mieux le faire,
où peut-être d’utiliser des a priori un peu plus informatifs afin de
restreindre légèrement l’espace de paramètres exploré.
pp_check(m_gros, type = "bars")
Using 10 posterior draws for ppc type 'bars' by default.
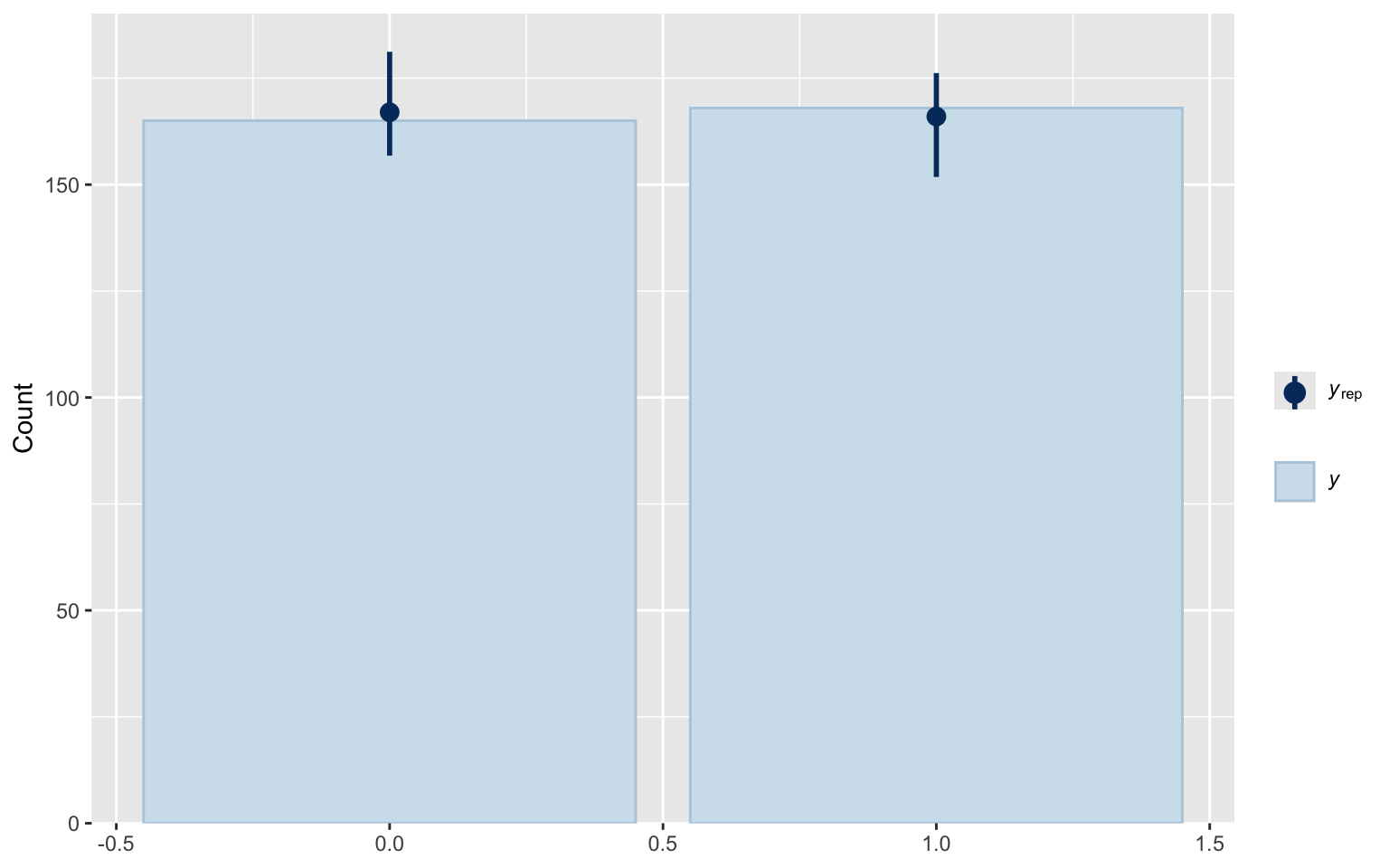
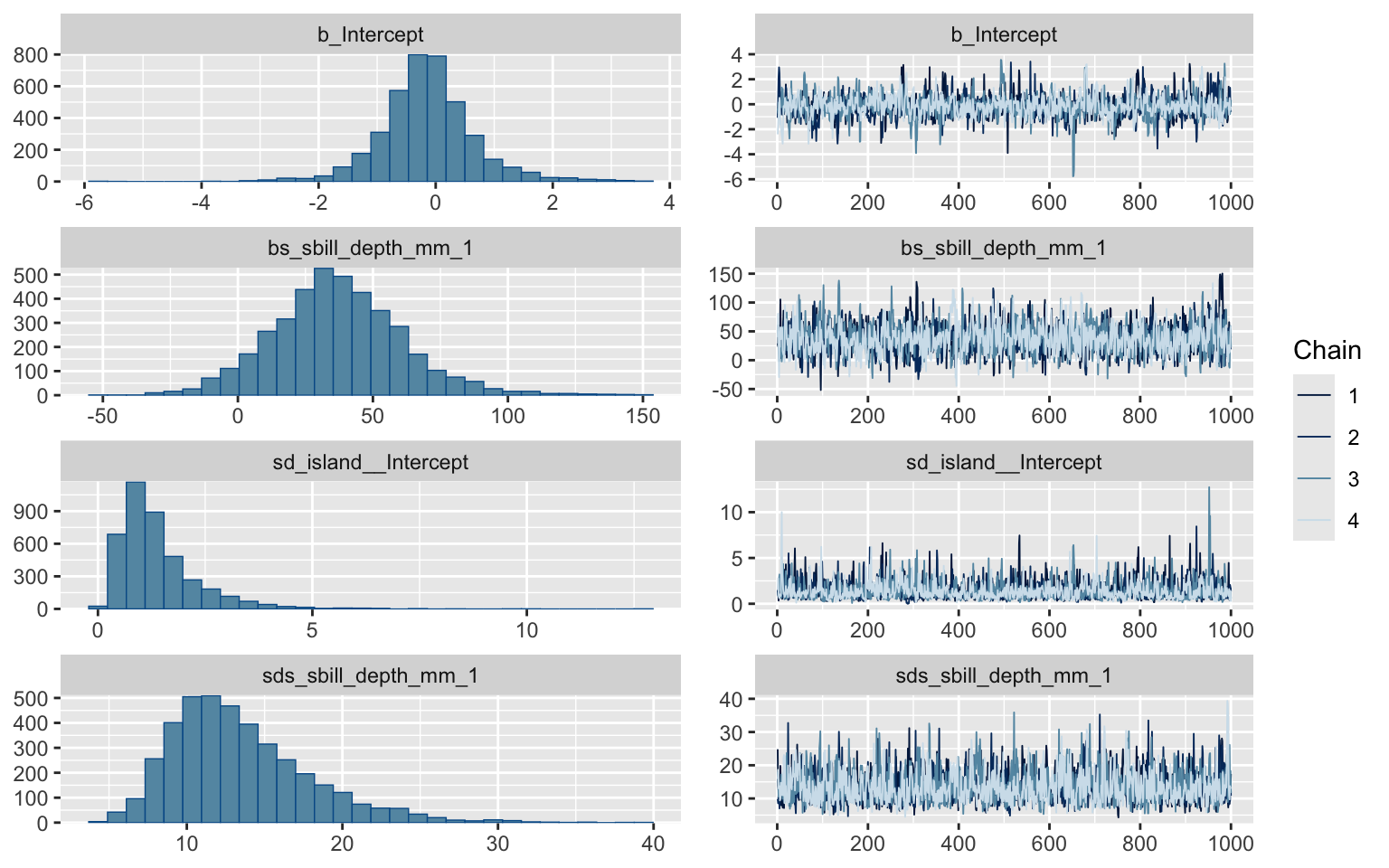
plot(m_gros)
loo(m_gros)
Computed from 4000 by 333 log-likelihood matrix.
Estimate SE
elpd_loo -164.6 10.8
p_loo 9.2 1.9
looic 329.2 21.7
------
MCSE of elpd_loo is 0.1.
MCSE and ESS estimates assume MCMC draws (r_eff in [0.7, 1.2]).
All Pareto k estimates are good (k < 0.7).
See help('pareto-k-diagnostic') for details.
summary(m_gros)
Family: bernoulli
Links: mu = logit
Formula: sex ~ s(bill_depth_mm) + (1 | island)
Data: pour_gros_modele (Number of observations: 333)
Draws: 4 chains, each with iter = 2000; warmup = 1000; thin = 1;
total post-warmup draws = 4000
Smoothing Spline Hyperparameters:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS
sds(sbill_depth_mm_1) 13.44 4.51 6.91 24.25 1.00 1748
Tail_ESS
sds(sbill_depth_mm_1) 2198
Multilevel Hyperparameters:
~island (Number of levels: 3)
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
sd(Intercept) 1.39 0.96 0.36 3.78 1.00 1259 1866
Regression Coefficients:
Estimate Est.Error l-95% CI u-95% CI Rhat Bulk_ESS Tail_ESS
Intercept -0.15 0.83 -1.72 1.67 1.00 1212 1253
sbill_depth_mm_1 37.01 24.50 -9.01 88.73 1.00 1867 1610
Draws were sampled using sampling(NUTS). For each parameter, Bulk_ESS
and Tail_ESS are effective sample size measures, and Rhat is the potential
scale reduction factor on split chains (at convergence, Rhat = 1).
L’autre chose pratique des adaptations du R2 proposées pour les modèles bayésiens est que ce dernier fonctionne aussi dans un GLMM :
bayes_R2(m_gros)
Estimate Est.Error Q2.5 Q97.5
R2 0.3713249 0.02979675 0.3059714 0.4229299
Et il peut être partitionné pour obtenir la partie expliquée uniquement par les effets fixes du modèle :
bayes_R2(m_gros, re_formula = NA)
Estimate Est.Error Q2.5 Q97.5
R2 0.3272364 0.03998257 0.2286067 0.3851488
Évidemment, ici, il faudrait en faire beaucoup plus pour valider la qualité du modèle. Entre autres, probablement explorer les taux de faux positifs, faux négatifs, etc.
Pour aller plus loin?
Si jamais vous voulez poursuivre votre parcours avec brms, je vous conseille de
consulter l’article original publié par l’auteur au moment de la sortie de la
librairie :
Bürkner, P.-C. (2017). brms: An R Package for Bayesian Multilevel Models Using Stan. Journal of Statistical Software, 80(1), 1–28. https://doi.org/10.18637/jss.v080.i01
Bürkner a aussi préparé une série de vignettes illustrant les plus importants cas de figures d’utilisation de la librairie : https://paulbuerkner.com/brms/articles/index.html
Enfin, surveillez bien dans les prochains mois, puisque Bürkner planche aussi sur un livre complet
détaillant les applications de brms. Une première version du livre est d’ailleurs déjà disponible sur son site web : https://paulbuerkner.com/software/brms-book/